



Activation of second messenger pathways in alveolar macrophages by endotoxin
- M.M. Monick and
- G.W. Hunninghake
- Dept of Medicine, University of Iowa College of Medicine and Veterans Administration Medical Centre, Iowa City, IA, USA
- M.M. Monick, Division of Pulmonary, Critical Care and Occupational Medicine, Room 100, EMRB, The University of Iowa Hospitals and Clinic, Iowa City, IA, 52242, USA. Fax: 1 3193356530. E‐mail: martha-monick@uiowa.edu
Abstract
The alveolar macrophage plays an important role in immune surveillance of the lung. Early responses to infectious agents by macrophages can decrease tissue injury and promote recovery of the host. Macrophage responses to pathogens are the cornerstone of the innate or nonspecific immune system. In particular, the response of macrophages to endotoxin from Gram negative bacteria has been the focus of many recent studies. The recent discovery of the endotoxin receptor has accelerated the study of signalling in macrophages. This review focuses on the downstream events that occur following exposure of the alveolar macrophage to endotoxin.
- endotoxin
- macrophage
- mitogen-activated protein kinase
- nuclear factor‐κB
- phosphatidylinositol 3‐kinase
- sphingolipids
Macrophages are the major effector cells of the innate immune system (fig. 1⇓). The term “macrophage” was first used more than 100 yrs ago by Elie Metchnikoff to describe large mononuclear phagocytes seen in tissues 1. Macrophages are highly distributed throughout the body in the lymphoid organs, liver, lung, gastrointestinal tract, central nervous system, serous cavities, bone, synovium and skin. They originate in the bone marrow as monoblasts, move into the blood stream as monocytes and finally move into the tissue and differentiate into resident macrophages 2.

Alveolar macrophages respond to endotoxin (LPS) by the generation of inflammatory mediators. These include growth factors, cytokines, chemokines, lipids and reactive gases. NO·: nitric oxide; O2·−: superoxide radical.
The innate immune system (as opposed to the targeted antimicrobial response of the adaptive immune system) is the organism's first line of defense against microbial invasion. It functions to decrease tissue injury and death, promote recovery of the host, decrease secondary or opportunistic infections and interact with the adaptive immune system 3–6. The principle components of the innate immune system include antimicrobial substances produced by epithelial cells, neutrophils, natural killer cells and macrophages. Macrophages are found in abundance at sites where organisms interact with the environment. Of the many types of macrophages found in the body, the alveolar macrophage has the most frequent contact with external stimuli. Each day, a human being breathes in and out more than 7,000 L of air 7. This leads to a continual influx of organic and inorganic dusts and bacteria. The early recognition and response to invading pathogens by the alveolar macrophage is essential to survival. This early interaction between macrophage and environment results in the generation of signalling cascades, leading to the various effector functions that make up the initial immune response in the lung (fig. 1⇑). This review focuses on the responses that occur following the interaction of endotoxin (LPS) from Gram-negative bacteria and macrophages, with a focus on the alveolar macrophage.
LPS is the principle activating component of the Gram-negative cell wall 8. It is a potent molecule and is known to activate macrophages down to a level of 1 nM 3. Janeway and Medzhitov 9, 10 have defined components of the innate immune system, and by their classification, LPS is a pathogen-associated molecular pattern (PAMP). The cellular receptors that interact with PAMPs are known as pattern recognition receptors (PRRs). LPS interacts with particular PRRs on the macrophage surface. LPS contains an acylated diglucosamine head group (Lipid A) linked to a chain of repeating disaccharides. The Lipid A portion is responsible for the biological activity of LPS, while recent evidence suggests that the polysaccharide tail determines the antigenic properties 11, 12. Interaction of LPS with a receptor complex on alveolar macrophages causes the sequential activation of multiple signalling pathways and transcription factors, resulting in gene transcription. This results in the orchestrated production of both pro- and anti-inflammatory mediators.
Endotoxin receptors
The major receptors found on the alveolar macrophage include scavenger receptors, Fc receptors, G protein coupled receptors (GPCRs), integrins, CD14, Toll-like receptors (TLRs), cytokine receptors and chemokine receptors 13–19. For LPS to interact with the macrophage it must first come into contact with LPS binding protein (LBP). LBP is required for an optimal LPS signal in macrophages. In in vitro experiments, LPS without LBP has only a fraction of its macrophage activating capabilities. LBP is a phospholipid transport protein that binds to Lipid A from LPS and presents it to receptor CD14. Shortly after the discovery of LBP 20–22, other studies 23, 24 identified CD14 as an important part of the LPS signalling pathway. Since that time, extensive biochemical and genetic data have confirmed an important role for CD14 in the macrophage response to LPS. Both LBP and CD14 knockout animals are partially resistant to the effects of LPS and to a lethal challenge by LPS 25, 26. CD14 is a membrane glycoprotein with a glycosylinositol (GPI) tail. It is present on alveolar macrophages and is also found as a soluble form that lacks the GPI tail 27–29. CD14 is known to bind LPS, but the fact that it is anchored to the membrane with a GPI tail and contains no intracellular signalling moiety suggests a more complicated signalling complex.
The search for a signalling LPS receptor led to the discovery of Toll receptors. The gene encoding Toll was discovered in the 1980s as a Drosophila gene that affected dorsal/ventral patterning in the embryo 30. A generation of mutant flies provided the information that this transmembrane protein was involved in the immune response to fungi 31, 32. Toll was shown to be part of a cascade, including spaetzle, Toll, tube, pelle and cactus, which controlled the production of antifungal peptides. Shortly after this, the first mammalian Toll receptor was described by Medzhitov et al. 33. It was found on a monocyte cell line and shown to activate the transcription factor nuclear factor‐κB (NF‐κB) and induce cytokine production. Mammalian Toll is part of a homologous family of TLRs. Based on similarities in the cytoplasmic portion of the molecule, at least 10 different TLRs have been identified 34–39. The initial description of human TLR by Medzhitov et al. 33 was followed by the observation that TLR2 was the CD14 associated LPS receptor 40, 41. These studies were weakened by later studies that identified TLR4 as the LPS-specific receptor. The link between TLR4 and LPS first appeared in a study showing that the genetic mutation responsible for LPS hyporesponsiveness in C3H/Hej mice was in the TLR4 gene 42. Since that time, an additional study in LPS hyporesponsive mice C57BL10/ScNCr and LPS hyporesponsive humans has linked TLR4 mutations to the hyporesponsive phenotype 43–47. Further in vitro evidence strengthened the link between LPS and TLR4, while at the same time showing that other TLRs are involved in responses to other bacterial products (TLRs 2, 1 and 6) and to bacterial deoxyribonucleic acid (DNA) (TLR9). Although it was not the LPS receptor, TLR2 has been linked to peptidoglycans from Gram positive bacteria, lipoteichoic acid, lipoarabinomannan from mycobacteria, and lipopeptides 3, 11, 48, 49. Both TLR4 and TLR2 have been found on alveolar macrophages, providing the entry point for a complex cascade of LPS signalling 50.
In addition to the TLR complex, a number of other membrane receptors have been proposed to play a role in LPS signalling. These include the scavenger receptor, β2‐integrins, a purinergic receptor (P2X7), moesin, triggering receptor expressed on myeloid cells (TREM‐1) and a recently described complex that includes heat shock proteins 90 and 70, complement receptor 4 and bone morphogenic protein 51–56. Early studies have suggested that high-dose LPS could signal through some of these other receptors independent of CD14 and TLR4. The TLR4-/- knockout data, however, suggests that the TLR4 receptor is essential for LPS signalling 43. One possible explanation for the different data is that these other receptors might function as co-receptors and amplify the response after an initial CD14/TLR4 interaction with LPS. Further studies are necessary to definitively establish whether CD14/TLR4 independent signalling is possible. Several previous studies have suggested a role for the integrin receptors in LPS signalling 55, 57–59. A recent paper by Perera et al. 57 has shown that peritoneal macrophages from CD11b (a major β2‐integrin chain in macrophages) knockout mice have a decreased LPS response (some cytokines, cyclooxygenase (COX)2 and interleukin (IL)-12 p35, and a slight decrease in p38 activation). Because both in vivo and in vitro, the TLR4 knockout effect is so much greater than the effect of the β2‐integrin chain deletion, this review focuses on signalling downstream of the TLR4 complex.
Proximal signalling
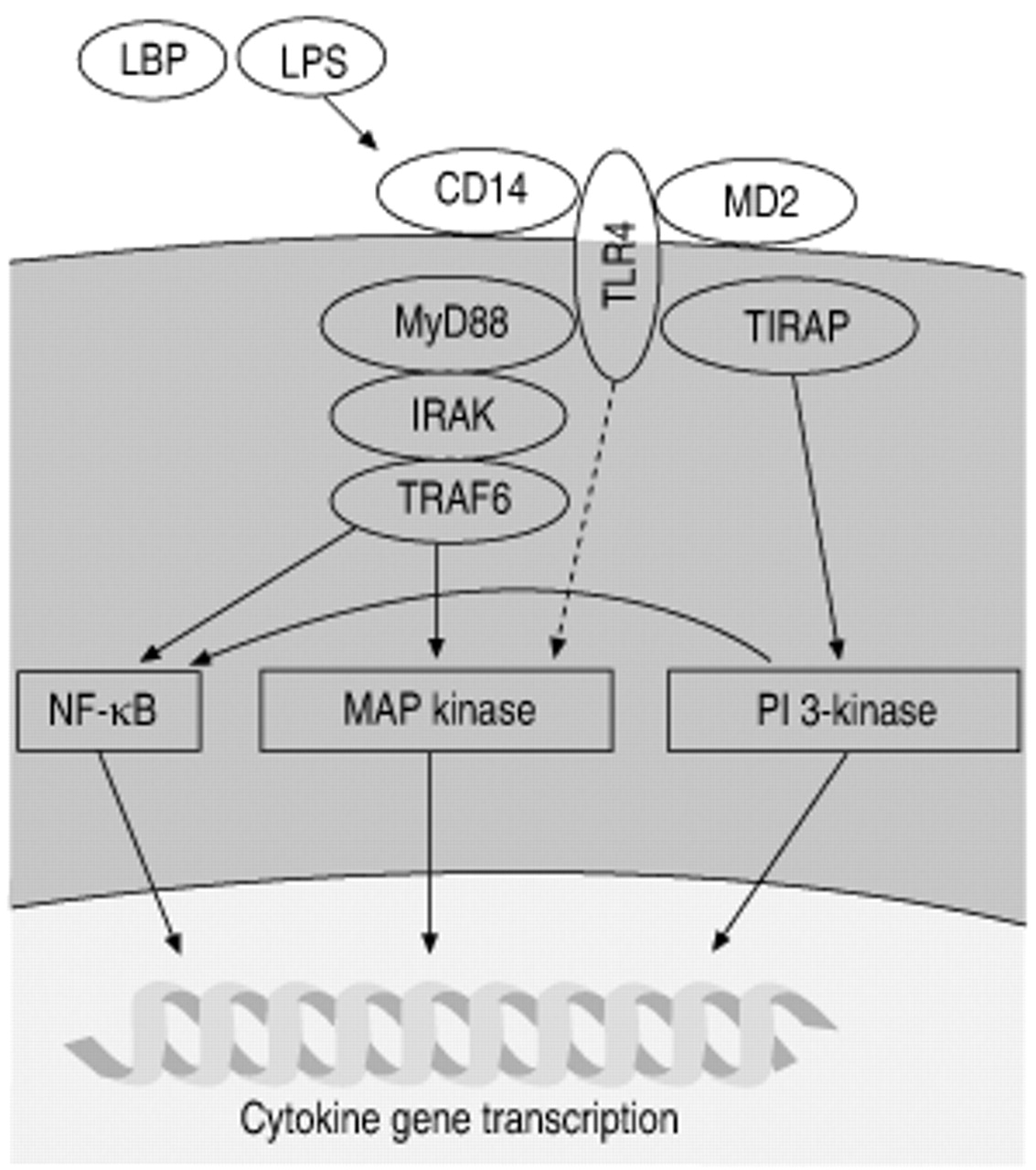
The cytoplasmic domain of all TLRs shares significant sequence homology with the IL‐1 and IL-18 receptors. This suggests a similar early signalling process after engagement of the receptor. The working model at this point is that LPS complexes with LBP, resulting in binding of CD14 and subsequent activation of TLR4 (with the assistance of a small secreted glycoprotein, MD‐2) (fig. 2⇓). Two recent studies have suggested a direct interaction of LPS and TLR4 12, 60. Downstream of TLR4, signalling resembles proximal IL‐1 signalling, which includes the sequential recruitment of the adaptor protein MyD88 and IL‐1 receptor-associated kinase (IRAK). Activated IRAK then returns to the cytoplasm where it activates tumour necrosis factor (TNF) receptor-associated factor 6 (TRAF6) 3, 6, 61. Signals downstream of TLR4 fall into two classes, MyD88 dependent and MyD88 independent (fig. 2⇓). Macrophages from MyD88-/- mice show the interesting phenotype of unimpaired or only decreased NF‐κB and mitogen-activated protein (MAP) kinase signalling and a total block of cytokine production 62. This suggests that there are important signalling pathways that are independent of MyD88. A new adaptor protein (Toll/interleukin 1 receptor homology domain (TIR) containing adaptor protein (TIRAP)) has been identified that provides MyD88 independent signalling from TLR4 63. After the initial activation of the TLR4 complex, a number of signalling pathways are activated. These include the MAP kinases, phosphatidylinositol (PI) 3‐kinase pathway, and sphingolipid metabolites.

TLR4 signalling Endotoxin (LPS) first binds to the receptor CD14. This interaction results in the presentation of LPS to Toll-like receptor 4 (TLR4). This, in turn, triggers the formation of a signalling complex that includes MyD88, Toll/interleukin (IL) 1 receptor homology domain containing adaptor protein (TIRAP), IL‐1 receptor associated kinase (IRAK), and tumour necrosis factor receptor-associated factor (TRAF) 6. Signalling downstream of this complex leads to activation of multiple pathways that regulate LPS-induced gene transcription. NF‐κB: nuclear factor‐κB; MAP: mitogen-activating protein; PI: phosphatidyinositol. LBP: LPS binding protein.
Mitogen-activating protein kinesis
The MAP kinases are a family of evolutionarily conserved enzymes that connect cell surface receptors to regulatory targets, which include both cytoplasmic and nuclear proteins. They can be activated by divergent stimuli, including hormones, growth factors, cytokines, GPCR agonists, tumour growth factor β and TLR ligands 64. They are activated by a three-tiered cascade of kinases: 1) MAP kinase kinase kinase (MKKKs), of which there are many and a redundancy of function (for example MAP kinase kinase 4 (MEKK4) can be phosphorylated by at least 10 different MKKKs); 2) a suset of MKKK (MEKKs), of which there are only a few per MAP kinase; 3) MAP kinases themselves. The three major MAP kinase families are the extracellular signal-related kinase (ERK) (1 and 2), p38 (α, β, γ, and δ) and the c‐Jun N‐terminal kinase (JNK) (1, 2 and 3) 64. All three of these are activated by LPS in alveolar macrophages. The MAP kinases are activated by phosphorylation of a threonine (Thr) X tyrosine (Tyr) conserved site in subdomain VII of the activation loop (ERK (Thr Glu Tyr), p38 (Thr Gly Tyr), and JNK (Thr Pro Tyr)) 65. All MAP kinases recognise similar phosphoacceptor sites composed of a serine or threonine followed by a proline. The amino acids surrounding this site determine the specificity 64. Activation of MAP kinases in alveolar macrophages is an early event after LPS contact, occuring within the first 15 min 66–69.
All three major MAP kinase families are activated by LPS in monocytic cells and all are involved in activation of transcription factors that are important for cytokine production 70–74. The link between MAP-kinase activation and the production of inflammatory mediators has been studied extensively (fig. 3⇓). The links to activator protein‐1 (AP‐1) shown in this figure complement the role of MAP kinases in NF‐κB activation.
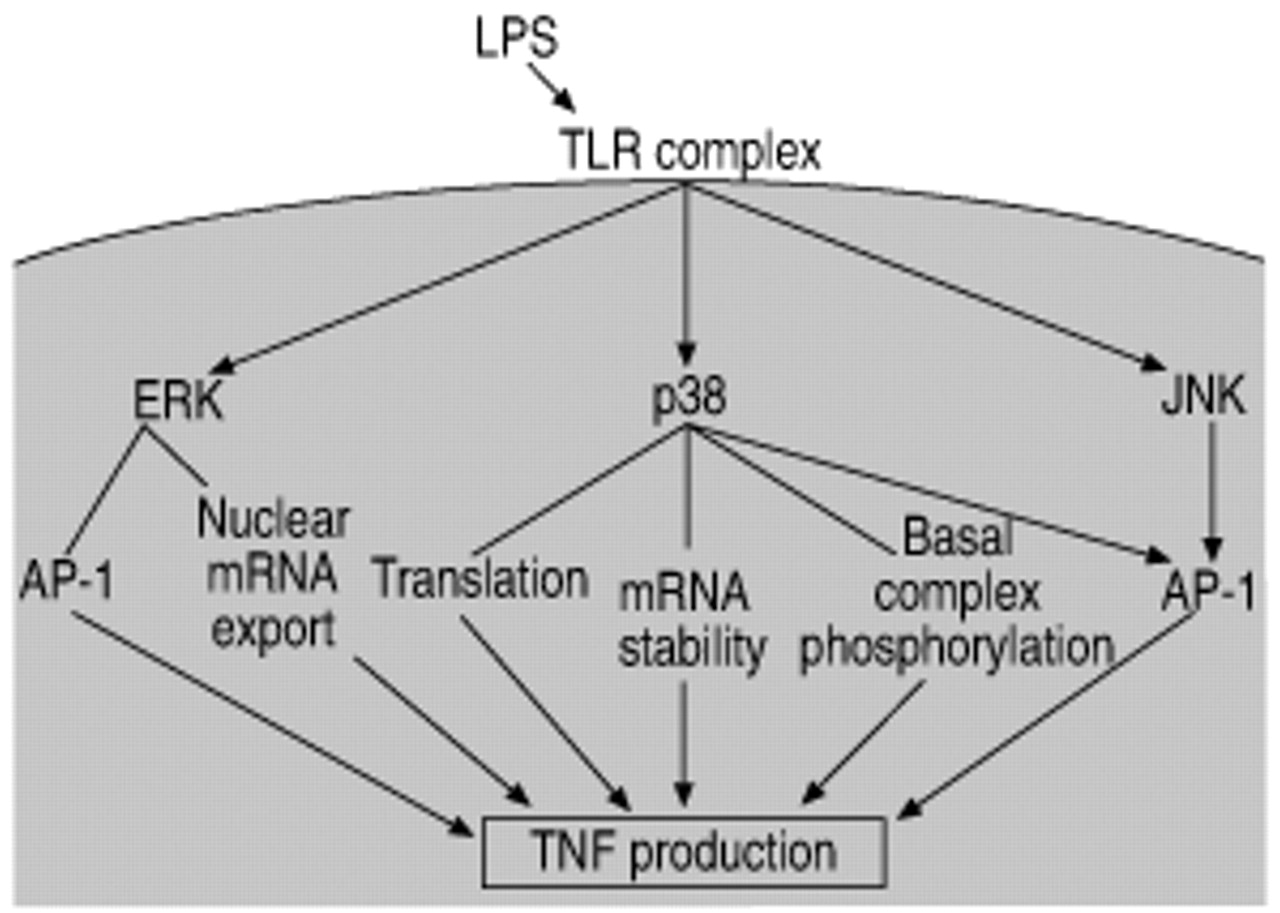
Mitogen-activating protein (MAP) kinases. MAP kinase activation by endotoxin (LPS) results in transcriptional activation of the tumour necrosis factor (TNF) gene and early release of TNF from the macrophage. Each of the MAP kinase pathways plays multiple and distinct roles in this event. TLR: Toll-like receptor; ERK: extracellular signal-related kinase; JNK: c‐Jun N‐terminal kinase; AP‐1: activator protein‐1; mRNA: messenger ribonucleic acid.
JNK was initially identified by Kyriakis and coworkers 75, 76 as a p54 microtubule-associated protein that was activated by cycloheximide. A major substrate of JNK is c‐Jun, a member of the AP‐1 transcription factor dimer. JNK binds to c‐Jun and phosphorylates it on serines 63 and 73 77. This phosphorylation is necessary for the transcriptional activity of c‐Jun (rather than the DNA-binding activity) 78. The involvement of MAP kinases (JNK-induced phosphorylation) in the transcriptional activity of a transcription factor as a process distinct from DNA binding is also found in studies on the activity of NF‐κB (see Transcription factors section). JNK also phosphorylates other AP‐1 proteins, including Jun B, Jun D, and activating transcription factor‐2 (ATF‐2) 79. While JNK phosphorylation of AP‐1 proteins plays a positive role in transcriptional activity, AP‐1 activity can also be negatively regulated by phosphorylation. Phosphorylation of c‐Jun at the DNA binding domain (threonine 231, threonine 239, serine 243 and serine 249) by glycogen synthase kinase (GSK‐3) or casein kinase 2 inhibits DNA binding 80, 81. In a study performed in LPS-treated alveolar macrophages, it was found that LPS inactivated GSK‐3 (a constitutively active kinase), suggesting that LPS regulation of AP‐1 activity involves both activation (JNK) and blocking of constitutive inhibition (GSK‐3) 82.
Direct p38 phosphorylation is linked with activity of the transcription factors ATF‐2, stress-activated protein kinase 1a (Sap1a) and myocyte enhancer binding factor (MEF) 2C family members 83. p38 can phosphorylate ATF‐2 at threonine 69 and serine 71 in the transcriptional domain, and this phosphorylation correlates with increased transcriptional activity 84. Sap1a is a member of the ternary complex factor (TCF) family and is phosphorylated by p38 on serines 381 and 387 85–88. The transcription factor MEF2C plays a role in c‐Jun transcription and is phosphorylated and activated by p38 83.
There are less direct links between ERK and transcription factor phosphorylation, although all of the MAP kinase family members phosphorylate and activate transcription factors of the ETS family that are involved in fos transcription. Phosphorylation of ELK‐1 (a member of the ETS family) is a defining characteristic of ERK activation 89. In addition, a recent paper by Young et al. 90 shows that ERK phosphorylates the AP‐1 protein Fra‐1 on threonine 231, suggesting involvement of ERK in the transcriptional activity of AP‐1.
MAP kinases phosphorylate both cytoplasmic and nuclear substrates. Nontranscription factor effects of p38 include increasing messenger ribonucleic acid (mRNA) stability and phosphorylation of basal transcription complex components. p38 involvement in TNF mRNA stability was found both in a study by Rutault et al. 91 and in a study examining the mechanism of IL-10 mediated suppression of LPS-induced TNF 92. A role for p38 in translational regulation of cytokine production was found in a study of MAP kinase activating protein (MAPKAP)‐2 (a kinase directly downstream of p38) knockout mice 93. In the MAPKAP‐2 knockout animals, LPS-induced septic shock resulted in 90% less TNF than in wild-type animals. However, the TNF mRNA levels were the same. The authors concluded that p38 and MAPKAP‐2 were essential components of the biosynthesis of TNF at the post-transcriptional level, possibly having a role in the translation process. Production of other inflammatory mediators supports the role of p38 in mRNA stability. A study by Dean et al. 94 demonstrates that p38 is essential for the stabilisation of COX2 mRNA.
Nontranscription factor effects of ERK have also been documented. In the cytoplasm, ERK has been shown to phosphorylate a diverse array of substrates: p90 ribosomal S6 kinase (p90RSK), cytosolic protein kinase A (PKA), tau, endothelial growth factor receptor (EGFR), Raf‐1 and fos 64. Tpl2 is a kinase that is necessary for ERK activation and Tpl2 knockout mice demonstrate a link between ERK kinase and TNF mRNA nuclear export 95. Without ERK activity, TNF mRNA remained trapped in the nucleus, blocking subsequent protein production. This study suggests that ERK is involved in the export of cytokine mRNAs from the nucleus.
Many studies have addressed the issue of how LPS activation of mononuclear cells results in the activation of MAP-kinase pathways, including the ERK pathway 66–69. The consensus pathway for ERK signalling has been defined as activation of the small GTPase Ras, which recruits the kinase Raf‐1, which then activates the dual kinase MEK1 or 2, which phosphorylates a threonine and tyrosine in ERK 1 or 2. In murine RAW264.7 macrophages, Geppert et al. 96 demonstrated that LPS activated ERK and that dominant negative constructs of Ras or Raf‐1 inhibited this activation as well as LPS-induced TNF production. A number of studies substantiated this observation, but not all studies supported a role for Raf‐1 in LPS signalling to ERK 69.
Studies like Geppert et al. 96, which used overexpression of dominant negative constructs, generally concluded that LPS signals via Raf‐1. In contrast, other studies have concluded that LPS activation of ERK is Raf‐1 independent 69, 97, 98. In a study performed by the current authors using normal human alveolar macrophages, LPS-induced ERK activation was found to be Raf‐1 independent. LPS activated the atypical protein kinase C (PKC) isoform ζ, and it was PKC ζ that activated MEK. The significance of an alternative pathway to ERK activation is currently being studied. It is possible that in alveolar macrophages, Raf‐1 is involved in alternative signalling. For example, Raf‐1 has recently been shown to interact with and inhibit the apoptosis signal-regulating kinase 1 (ASK‐1) 99. This action of Raf‐1 is independent of MEK and ERK.
LPS signalling to cytokine genes involves MAP kinases other than ERK. This was shown in a study by Hambleton et al. 100, which used a cell line with a Raf‐1 chimeric protein that can be activated for prolonged time periods. With prolonged ERK activation there was very little production of TNF (20 times less) compared to LPS-treated cells, suggesting the need for pathways other than ERK for LPS-induced TNF production. The current authors found that in alveolar macrophages, optimal cytokine signalling (TNF and IL‐6) involved both ERK and p38 MAP kinases 66. If either ERK or p38 signalling were blocked with specific inhibitors, a partial block of cytokine transcription was found. If both inhibitors were used, there was a complete block. This has been confirmed in a recent study by Rutault et al. 91 who found that optimal TNF production required both ERK and p38. They also found that p38 was linked to an increase in TNF mRNA stability 91.
LPS has also been shown to activate the JNK MAP kinase 71, but less work has been done on the exact role of JNK in LPS signalling. The direct link between JNK and phosphorylation of the proteins involved in AP‐1 generation links this kinase to cytokine production and further studies will no doubt refine this knowledge.
Phosphatidylinositol 3‐kinase pathway
Until the 1970s lipids were considered inert structural parts of cell membranes. This began to change with discovery of the phosphoinositide cycle 101, 102. Since that time, signalling functions for multiple classes of lipids, including phosphoinositides and arachidonic acid metabolites, have been demonstrated. A lipid signalling pathway with a definitive role in LPS signalling is the PI 3‐kinase pathway (fig. 3⇑) 82, 103. Membrane-associated PI 3‐kinase catalyses the transfer of adenosine trisphosphate to the D‐3 position of the inositol ring of membrane-localised phosphoinositides 104. This results in the production of a number of bioactive species including phosphatidylinositol 3 phosphate (PI3P), phosphatidylinositol 3,4 phosphate (PI3,4P), and phosphatidylinositol 3,4,5 phosphate (PI3,4,5P). Both PI3,4P and PI3,4,5P are nominally absent in most unstimulated cells and increase dramatically following PI 3‐kinase activation. The production of PI3,4,5P, especially, results in the recruitment of 3‐phosphoinositide-dependent kinase (PDK‐1), a kinase with multiple downstream substrates, including protein kinase B (Akt) (fig. 4⇓) 105.

Activation of phosphatidylinositol (PI) 3‐kinase results in a signalling cascade that begins with the production of phosphatidylinositol 3,4,5. This leads to the membrane recruitment of 3‐phosphoinositide-dependent kinase (PDK)‐1 and other downstream kinases. PDK‐1 phosporylates these substrates in their activation domains generating active kinases. These kinases then phosphorylate further substrates, amplifying the initial PI 3‐kinase activity several times. Akt: protein kinase B; PKC: protein kinase C; PKA: protein kinase A.
Akt, like PDK‐1, is recruited to membrane-bound D‐3 phosphorylated phosphatidylinositols (D‐3 PPIs) by its pleckstrin homology (PH) domain. Binding of Akt to D‐3 PPIs results in a conformational change allowing phosphorylation by PDK‐1 (on threonine 308 in the activation loop) and an activating phosphorylation at serine 473 within the hydrophobic motif at the kinase tail 106. Activation of Akt results in the phosphorylation of a number of substrates that have potential importance in LPS signalling (GSK‐3, Bad, caspase 9, Forkhead transcription factors, Raf‐1, inhibitor of κb (IκB) kinase, phosphodiesterase (PDE)‐3B and endothelial nitric oxide synthase (eNOS)) 104, 106, 107. Phosphorylation of these proteins by Akt results in either activation or inactivation depending on the substrate. Inactivation of some of the pro-apoptotic factors (caspase 9, Bad, GSK‐3 and the Forkhead family of transcription factors) is central to Akt's role in cell survival 108–111.
Activation of PI 3‐kinase was first described as being downstream of tyrosine kinases. SH2 domains in the p85 regulatory unit of Class I PI 3‐kinase were found to bind to phosphotyrosines in the cytoplasmic domain of growth factor receptors initiating signalling 112. This results in the membrane recruitment of the p110 catalytic unit and subsequent signalling. LPS activation of PI 3‐kinase was first described by Herrera-Velit et al. 113. They found that in monocytes, LPS caused a rapid activation of PI 3‐kinase that was accompanied by the production of D‐3 PPIs. A second study by the same group showed that the LPS-induced PI 3‐kinase activity was linked to activation of PKC via PDK‐1 114. This is consistent with the findings of the current authors in alveolar macrophages when ERK activation was linked to PKC ζ 69. It was found that LPS caused an association between PKC ζ and PDK‐1 that temporally coincided with activation. Since that time, two further studies in alveolar macrophages examining activation of PI 3‐kinase by LPS have been completed. In the first of these studies, the current authors showed that LPS activated PI 3‐kinase and the downstream kinase Akt 82. This resulted in a link to GSK‐3 inactivation and the nuclear accumulation of β catenin. β Catenin driven genes include cyclin D1, c‐myc, fra 1 and connexin 43. Both activation of Akt and inactivation of GSK‐3 are linked to cell survival and next a link between LPS activation of PI 3‐kinase and cell survival in alveolar macrophages was demonstrated 103.
Having shown some of the events downstream of LPS induced PI 3‐kinase activity, the current authors then evaluated the link between the TLR complex and activation of PI 3‐kinase. A group at Scripps Research Institute (La Jolla, CA, USA) looking at the link between TLR2 (Gram-positive signalling) and PI 3‐kinase activation 115 found a p85 binding site on the cytoplasmic domain of TLR2. They linked TLR signalling to direct recruitment of p85 to the receptor and activation of PI 3‐kinase. TLR4 lacks this domain and in alveolar macrophages it was found that LPS signalled PI 3‐kinase via the generation of ceramide. In addition, it was found that the ceramide-induced PI 3‐kinase activation masked the normal apoptotic function of ceramide and turned what could have been a cell death-inducing event (the generation of ceramide) into a non-apoptotic event (activation of ERK and PI 3‐kinase with its downstream kinases). In addition to regulating cell survival, the PI 3‐kinase dependent kinase, Akt, is linked to activation of the transcription factor NF‐κB. This will be dealt with more completely in the section on LPS regulation of the major cytokine-linked transcription factors.
Sphingolipids
The authors' studies in alveolar macrophages have demonstrated a link between LPS-generated ceramide and activation of both the ERK MAP kinase and PI 3‐kinase. Ceramide is only one of many sphingolipids that have potential signalling capacity, but it is the one that has the strongest demonstrated link with LPS signalling. Sphingolipids are defined by the presence of a sphingoid backbone (fig. 5⇓) 116. Ceramide is sphingosine acylated at the 2‐amino acid position. Sphingolipids are synthesised in the endoplasmic reticulum (ER) and the golgi. Ceramide can be generated by de novo synthesis initiated with the condensation of serine and palmitoyl-CoA, catalysed by serine palmitoyltransferase and ending with the conversion of dihydroceramide to ceramide by dihydroceramide reductase 116. Ceramide produced in the ER is moved to the golgi, where sphingomyelin synthase transfers the phosphocholine headgroup from phosphatidylcholine to the hydroxy group of ceramide, producing sphingomyelin. Sphingomyelin then moves to the plasma membrane where most is found on the outer leaflet. More particularly, sphingomyelin is especially prevalent in rafts or caveolae, plasma membrane structures that have been linked to signalling cascades 117. Some macrophage receptors have been localised to caveolae (CD14 and GPCR, for example), possibly linking LPS signalling to alterations in the lipid component of macrophage membranes.

Sphingolipid metabolism. A diagram of a possible ceramide-generated sphingolipid pathway in alveolar macrophages. Ceramide can be rapidly converted to sphingosine and sphingosine 1 phosphate and just as rapidly converted back depending on the presence and activity of the converting enzymes. SIP: sphingosine 1 phosphate.
Ceramide involved in signalling is not generated by de novo synthesis but rather triggered within minutes via the action of neutral and acid sphingomyelinases 116, 118, 119. These enzymes convert sphingomyelin (on the plasma membrane or on lysosomal membranes) to ceramide (fig. 4⇑). Sphingomyelinase-generated ceramide is found after cellular exposure to a diverse array of stimuli, including TNF‐α, Fas ligand, dexamethasone, nitric oxide, IL‐1β, interferon γ and LPS 116. Many of these events are pro-apoptotic. LPS-induced ceramide in alveolar macrophages is one of the few places that ceramide generation has been linked to cell survival 103. How ceramide activates signalling cascades has not been clearly defined. Two possible downstream mediators are ceramide-activated proline-directed kinase 120 and ceramide-activated protein phosphatase 116. Other possible links include novel and atypical PKC isoforms, c-Raf and protein phosphatase 1B (PP1B) 121.
Other sphingolipids that may be involved in LPS signalling include sphingosine, generated by the actions of ceramidases on ceramide, and sphingosine 1 phosphate, generated by the actions of sphingosine kinase on sphingosine (fig. 4⇑). Sphingosine was first described as an inhibitor of PKC 122. This was an in vitro observation and there is some question about how relevant this is to the in vivo system 123. Sphingosine has been shown to mediate cell growth arrest by dephosphorylating retinoblastoma (Rb), and increases in sphingosine are linked to apoptosis in many systems 124, 125. In contrast, sphingosine 1 phosphate has been linked to cell survival 126–128. While a number of studies, including those of the current authors, have linked LPS to the generation of ceramide in macrophages 68, 69, 103, 129–131, there is as yet no data on the possible role of these ceramide metabolites in LPS signalling. This is likely to provide a fruitful area of research in the future.
Transcription factors
The end result of the signalling generated by LPS in macrophages is activation of transcription factors and the resulting production of both pro- and anti-inflammatory mediators. This review focuses on the transcription factor NF‐κB because of the significant role it plays in the production of many inflammatory mediators. NF‐κB activation has been linked to production of TNF‐α, IL‐1β, IL‐6, IL‐8, COX2, intracellular adhesion molecule (ICAM)‐1 and collagenase, to name just a few of the genes with active NF‐κB sites 132. LPS was first shown to activate NF‐κB in monocytic cells in a study by Muller et al. 133. Since that time, NF‐κB regulation after LPS has been the subject of intense study. What has emerged from these studies is the idea that there are multiple regulatory pathways involved in generating NF‐κB driven transcription. Regulation can be divided into four different areas: nuclear translocation, phosphorylation of Rel family proteins, interaction with the basal transcription complex and redox regulation.
The NF‐κB/Rel family of genes includes p50, p105, p52, p65 (Rel A), Rel B, and c‐Rel. The primary NF‐κB dimer involved in LPS signalling is a p65/p50 heterodimer 134, 135. NF‐κB proteins reside in the cytoplasm in an inactive state, kept there by IκBs. The IκB family includes α, β, and ε isoforms and the α isoform has been most closely linked to LPS signals 136. Phosphorylation of IκB by IκB kinases (IKKs) results in the ubiquitination of IκB. IκB is then degraded by the 26S proteosome releasing NF‐κB proteins to move to the nucleus 137. The IKK complex includes IKKα, β and a structural/regulatory component, γ 138. IKKβ has been identified as the primary IKK involved in LPS activation of NF‐κB in monocytic cells 139–141. Phosphorylation and activation of IKKβ by an upstream kinase leads to NF‐κB localisation in the nucleus (fig. 6a⇓). Looking upstream of IKKβ, there have been a number of proteins identified as possible IKKs. These include NF‐κB inducing kinase (NIK), transforming growth factor‐β‐activating kinase (TAK) 1, MEKK1, MEKK2, MEKK3 and Akt (a member of the PI 3‐kinase pathway) 142–144. Swantek et al. 141 demonstrated that negative mutants of NIK, TAK1 and Akt blocked LPS activation of NF‐κB in murine RAW264.7 cells. Several studies have defined NIK as the upstream kinase responsible for NF‐κB activation, but studies in alymphoplasia (ala) mice, which contain a disrupting point mutation in NIK, show no effect on NF‐κB activation by TNF 145. This has led to an increased interest in defining IKKs. One recent study has linked TRAF6, from the TLR pathway, to evolutionarily conserved signalling intermediate in Toll pathways (ECSIT)-MEKK1-IKKβ 146. Another possible TRAF6 signalling sequence is TRAF6 to TAK1-binding protein (TAB) 2‐TAK1-IKKβ 147.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Nuclear factor‐κB (NF‐κB) activation is regulated at multiple levels. a) Events that lead to the nuclear translocation of NF‐kB. b) Events that regulate the transcriptional activity of NF‐kB once it reaches the nucleus. LPS: endotoxin; TLR: Toll-like receptor; MEKK: a subset of mitogen-activating protein kinase kinase kinase; NIK: NF‐κB inducing kinase; TAK: transforming growth factor‐β‐activating kinase; Akt: protein kinase B; IKK: inhibitors of κB (IκB) kinases; PI 3: phosphatidylinsositol 3; mRNA: messenger ribonucleic acid; CBP: cAMP response element binding protein; PKA: protein kinase A.
Nuclear localisation is only one of the events that needs to happen for successful NF‐κB driven gene transcription. Several studies have shown that p65 must be phosphorylated for transcriptional competence (fig. 6b⇑). An early study by Cobb et al. 148 showed that a phosphocholine specific phospholipase C (PC-PLC) inhibitor could block NF‐κB without blocking transcription. A study by Yoza et al. 149 demonstrated that tyrosine kinase inhibitors also blocked transcription without blocking translocation. Finally, a number of studies have linked MAP kinases to transcriptional activity independent of translocation 66, 89, 150, 151. Possible upstream kinases of p65 include PKA, the atypical PKC isoform ζ and Akt 150, 152, 153.
Translocation and phosphorylation provide one level of regulation. A third level is redox regulation. In 1992, Matthews et al. 154 showed that NF‐κB DNA binding was inhibited by SH modifying agents and enhanced by reducing agents. Thioredoxin (an oxidoreductase with antioxidant properties) was identified as the protein that modified a cysteine residue in the p50 NF‐κB subunit, which allowed DNA binding. A more recent study has shown that thioredoxin plays a dual and opposing role in NF‐κB regulation. In cytosol, antioxidants, including thioredoxin, decrease activation of IKKβ and NF‐κB translocation, but after cell activation, thioredoxin moves to the nucleus where it plays a positive role in NF‐κB activity 155. These studies have mostly been done with stimuli other than LPS and with cells other than macrophages.
Besides thioredoxin, another protein involved in redox regulation of transcription factors is Ref‐1. Redox regulation of AP‐1 proteins by Ref‐1 is necessary for optimal DNA binding and transcriptional activity of AP‐1. The current authors performed a study in alveolar macrophages evaluating redox regulation of the transcription factor AP‐1 (also activated by LPS and involved in cytokine regulation) 156. Alveolar macrophages were deficient in Ref‐1 and this deficiency resulted in decreased AP‐1 DNA binding after macrophage activation. In addition, it was found that the growth factor granulocyte-macrophage colony-stimulating factor (GM-CSF) upregulates amounts of Ref‐1 in alveolar macrophages. This leads to increased AP‐1 DNA binding after stimulation of alveolar macrophages 157. The exact role played by thioredoxin and Ref‐1 in LPS signalling in alveolar macrophages needs further study.
A final point of regulation of NF‐κB‐driven transcription is the need for NF‐κB binding to members of the complex at the transcription start site. Necessary binding of NF‐κB to co-activators has been shown for the transcription factor cyclic adenosine monophosphate response element binding protein (CBP)/p300 and for the two transcription complex proteins TATA binding proteins (TBP) and transcription factor IIB (TFIIB) 66, 89, 153, 158, 159. It has been shown that LPS-induced NF‐κB transcription (a distinct form of DNA binding or p65 phosphorylation) requires phosphorylation of TFIID (TBP) via activation of the p38 MAP kinase pathway (fig. 6b⇑) 66, 89. The authors found that optimal NF‐κB transcription after LPS required p38-dependent phosphorylation of TBP. Inhibition of p38 had no effect of NF‐κB translocation or DNA binding but it did block phosphorylation of TBP. This study demonstrates that LPS-induced p38 activation plays a role in the regulation of NF‐κB activity via phosphorylation of a protein in the basal transcription complex.
Termination of NF‐κB signalling is a complex event. A large role is played by the rapid resynthesis of IκBα 135. Blocking of IκB transcription in LPS-treated cells results in the accumulation of NF‐κB 160. When LPS-induced IκB transcription is allowed to proceed, IκBa then binds to NF‐κB in the nucleus and the IκB/NF‐κB complex shuttles back to the cytoplasm via a nuclear export signal in IκB. LPS activation of NF‐κB in alveolar macrophages is a rapid process. The authors have found nuclear localisation as early as 30 min, with a peak of 1–3 h. Release of TNF happens quickly and is followed shortly by other inflammatory mediators. The process that shuts down NF‐κB activation in LPS treated alveolar macrophages involves the resynthesis of IκBα, but other mechanisms may arise from further studies.
Conclusion
Endotoxin signalling in alveolar macrophages results in a complex cascade of events resulting in the rapid production of first pro-inflammatory mediators and then more slowly, the production of mediators that slow or stop the inflammatory process. Activation of the innate immune system by endotoxin provides a first line of host defense ultimately controlling the initiation and determination of the adaptive immune response that follows. Significant advances have been made in understanding the pathways that lead to the macrophage response but many things remain to be learned.
- Received January 16, 2002.
- Accepted March 5, 2002.
- © ERS Journals Ltd