





Abstract
This is the first Australian study to examine survival and clinical characteristics in biopsy-proven idiopathic interstitial pneumonia.
A cohort of 70 patients from a single institution between January 1990 and December 1999 was reviewed.
All patients were Caucasian, 23 (33%) female. Mean age±sd at diagnosis was 60±12 yrs for males and 54±14 yrs for females. A total 24% of patients had never smoked. The histopathological diagnoses were usual interstitial pneumonia (UIP) (n=59), nonspecific interstitial pneumonia (NSIP) (n=7), desquamative interstitial pneumonia (n=3) and acute interstitial pneumonia (n=11). Clinical and functional characteristics of the two main histological subgroups of UIP and NSIP showed significantly older patients in the UIP group and a significantly lower mean forced expiratory volume in one second (FEV1) in the NSIP group. Median survival for UIP was 78 months compared with 178 months for NSIP. No survival difference between treated and untreated patients with UIP was found. Multivariate analysis revealed smoking alone to be predictive of poorer survival.
This study demonstrates the best median survival for usual interstitial pneumonia of available series and confirms a survival difference between usual interstitial pneumonia and nonspecific interstitial pneumonia. Furthermore, the reported results may have implications for treatment timing using conventional protocols currently recommended.
Idiopathic interstitial pneumonia is a term that encompasses the following entities: usual interstitial pneumonia (UIP), desquamative interstitial pneumonia (DIP), acute interstitial pneumonia (AIP), and nonspecific interstitial pneumonia (NSIP) 1. Several recent studies have demonstrated variable treatment responses and survival based on this classification 2–8. Formerly, lack of distinction between the various classes had led to them being considered together as idiopathic pulmonary fibrosis (IPF). The American Thoracic Society, in its International Consensus Statement 1 has now revised the definition of IPF as characterized solely by the histopathological pattern of UIP. This definition has now been accepted on an international level 9.
The aims of this study were to examine survival and clinical characteristics in patients identified as having idiopathic interstitial pneumonia in the context of their histological diagnosis.
Methods
A total 570, consecutive hospital medical records at a single institution were examined for patients diagnosed with pulmonary fibrosis between January 1990 and December 1999 (ICD9 codes 515 and 516.3). Patients with connective-tissue disease, occupational-dust exposure, other dust exposure, potential fibrogenic or antifibrogenic drug exposure or a syndrome associated with pulmonary fibrosis (e.g. neurofibromatosis) were excluded. All patients were human immunodeficiency virus (HIV) negative. A total 112 patients had clinical, physiological and radiographical criteria for IPF, in addition to having histological material available (transbronchial lung biopsy, open lung biopsy or autopsy).
Three pulmonary pathologists reviewed all histopathological material by consensus, unaware of clinical findings or previous histological diagnoses. Classification was undertaken using standard criteria for identifying UIP, NSIP, DIP and AIP 2, 4, 10. UIP is characterized by temporal and spatial heterogeneity with patchy lung fibrosis, areas of honeycomb change, fibroblastic foci and intervening normal lung parenchyma. NSIP, on the other hand, is a uniform appearing, cellular interstitial pneumonia typified by an interstitial lymphoplasmocytic infiltrate and accompanying amounts of interstitial fibrosis 4. No distinction was made in this study between the fibrosing and cellular patterns of NSIP according to the criteria described by Katzenstem and Fiorelli 5. In DIP the predominant abnormalities are within the alveolar spaces. There is a diffuse increase in intra-alveolar macrophages with only mild interstitial widening by uniform fibrosis and a chronic inflammatory cell infiltrate. The single case of AIP was characterised by changes consistent with acute and organising diffuse alveolar damage.
In total 42 biopsies were judged by the panel of pathologists to be inadequate for a definite diagnosis and these patients were excluded from the study. This included material consisting entirely of honeycomb change (end-stage lung) in which the underlying pathological process could not be determined.
The medical records of the remaining 70 patients meeting study criteria were reviewed. Data regarding age at diagnosis, sex, smoking history, occupation, family history, symptoms and their duration, signs at presentation, comorbidities, medications, laboratory investigations, arterial blood gas, respiratory-function test and exercise-test results, treatment and outcomes were collated. Respiratory function (spirometry, lung volumes and gas transfer) was measured on the Jaeger Compactlab Transfer and Body Systems (Jaeger, Hoechberg, Germany). Predicted values for the population were based on the following sources: Morris et al. 11 for spirometry, Cotes and Hall 12 for gas transfer, Goldman and Becklake 13 for lung volumes in females and Grimby and Sodenholm 14 for lung volumes in males.
A radiologist and clinician reviewed the radiographs (n=70) and computed tomography (CT) scans (n=52) together and agreed by consensus to classify the abnormalities as ground glass opacities, fine reticular-reticulo-nodular markings, ill-defined patchy infiltrates and honeycombing 15, 16. CT scans were obtained using the Elscint Twin Real Time Scanner (Elscent Ltd, Isreal) and chest radiographs, using Fuji computer radiography (FCR 9501 ES Energy Subtraction; Fujifilm Medical Systems USA Inc., Stamford, CT, USA). CT scans were used, where available, only to confirm the radiographical findings.
A mailed request for survival data was sent to all medical practitioners caring for patients who had not continued follow-up at the hospital. Minimum follow-up time was 1 month. Six patients, all with UIP, were lost to follow-up at this time. All had been confirmed as alive at 1 month. All were male. No other significant differences were found across all parameters examined between the group lost to follow-up and the group remaining. Survival was calculated from the date of the lung biopsy, which was also taken as the date of diagnosis. Deaths from any cause were included.
Statistical analysis involved two-sided tests with p-values <0.05 considered significant. Group comparisons were made using unpaired t-tests (for normally distributed continuous variables). A Wilcoxon's rank-sum test was used for non-normally distributed variables. Chi-squared statistics or Fisher's exact test were utilized for comparison of proportions. Cumulative survival probabilities were estimated using the Kaplan-Meier method. The log-rank test was used to compare survival of groups of patients. Univariate and multivariate analyses were conducted with reference to patient survival.
Results
All 70 patients with biopsy-proven idiopathic interstitial pneumonia were Caucasian; 23 (33%) were female. Mean age±sd at diagnosis was 60±12 yrs (range 16–88 yrs) for males and 54±14 yrs (range 30–82) for females. Twenty-four (34%) patients had never smoked. Twenty-nine (41%) patients were on medication for associated comorbidities. None of these medications were identified as having fibrogenic or antifibrogenic potential.
The histopathologic diagnoses included UIP (n=59), NSIP (n=7), AIP (n=1), DIP (n=3). The latter four patients will not be discussed further as their numbers were insufficient. The six patients with UIP lost to follow-up at 1 month were not included in subsequent analyses presented in this paper, apart from the survival analysis.
The clinical characteristics of the two main histological subgroups identified in this study UIP (n=53) and NSIP (n=7) are presented in table 1⇓. In the UIP group, a higher proportion of the patients were males compared with the NSIP group. The UIP patients were significantly older. Also, patients with NSIP had a significantly lower FEV1 compared to those with UIP (table 2⇓). However, one patient was known to have the copathology of emphysema.
Clinical characteristics of patients with usual interstitial pneumonia (UIP) and nonspecific interstitial pneumonia (NSIP)
Pulmonary function of patients with usual interstitial pneumonia (UIP) and nonspecific interstitial pneumonia (NSIP)
Radiographical patterns for the UIP group showed predominantly fine reticular markings (68%), followed by honeycomb changes (26%), ill-defined patchy infiltrates (9%) and ground glass opacities (7.5%). In the NSIP group, fine reticular markings were present in 71% of radiographs, with ill-defined patchy infiltrates in 43% and ground glass and honeycombing in 14%.
Other investigations performed in both groups of patients included erythrocyte sedimentation rate levels whose mean level was 30±18 in 25 patients with UIP and 21±16 in the seven patients with NSIP. Of 12 patients with UIP who had perinuclear antineutrophil cytoplasmic antibody levels performed, three had a recordable titre <1:640. Only one patient with NSIP had the levels examined with a negative result. Eighteen of 35 patients with UIP (seven were female) had low level antinuclear antibody (ANA) positivity, not exceeding titres of 1:640 as did two of seven patients with NSIP. No features of connective-tissue disease were noted either clinically or historically in any of these patients. No patient had antidouble stranded deoxyribonucleic acid antibodies detected in serum.
The histological material for diagnosis was obtained by open lung biopsy as the procedure of choice in the majority of patients with UIP and NSIP (table 3⇓).
Diagnostic procedures in patients with usual interstitial pneumonia (UIP) and nonspecific interstitial pneumonia (NSIP)
No deaths had been recorded in either group at 30 days postbiopsy. It is evident that the transbronchial biopsy was insufficient to allow for histological diagnosis in all except four cases and therefore a further procedure was required in 13 patients. Two patients were diagnosed only at autopsy.
The majority of patients received treatment, with most undergoing a trial of steroids (table 4⇓). Cyclophosphamide and cyclosporine were classified as cytotoxic drugs. Either one or the other was prescribed in the individual patient with only three UIP patients receiving cyclosporine during the course of their treatment. Records contained insufficient detail of treatment duration and drug dosage in the majority of cases to allow for further meaningful analysis.
Treatment modalities in patients with usual interstitial pneumonia (UIP) and nonspecific interstitial pneumonia (NSIP)
Follow-up data for survival were obtained for all patients (all six UIP patients lost to further follow-up were known to be alive at 1 month). Thirty patients (43%) were known to be deceased by December 31, 1999.
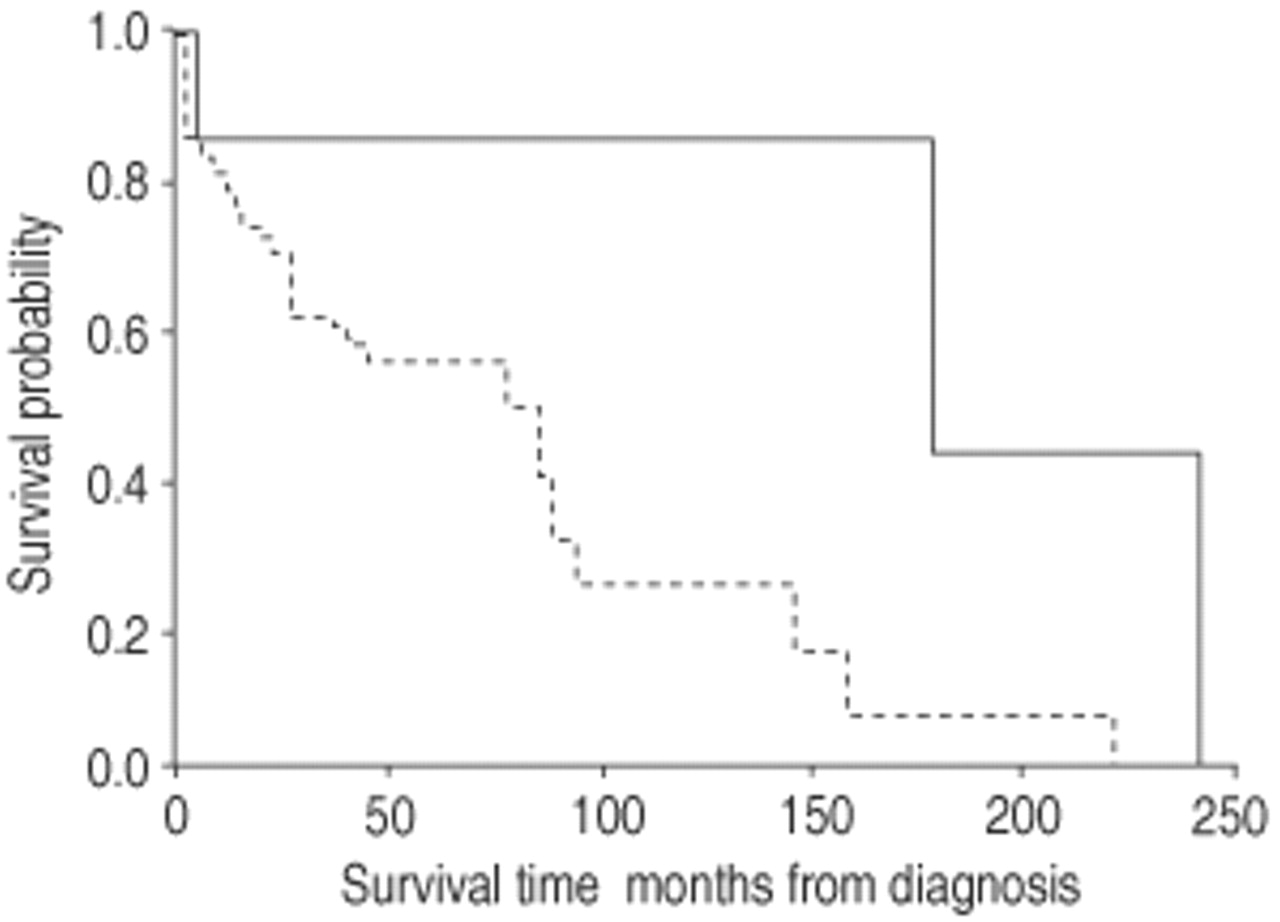
There was a significant difference in survival between patients with U1P and NSIP (p=0.03, log-rank test) (fig. 1⇓). Patients with UIP had a median survival of 78 months compared with 178 months for NSIP. For the UIP group, survival rates were 81% at 1 yr, 70% at 2 yrs, 58% at 5 yrs and 25% at 10 yrs. No significant difference was found in survival in the UIP group between those who were treated with medication (steroid/cytotoxic/colchicine) at some point in their management (n=28) compared to those who received no treatment (n=8) (p=0.14, log-rank test) (fig. 2⇓). Data were incomplete for 17 patients regarding treatment duration.

Survival in patients with usual interstitial pneumonia (- - -) and nonspecific interstitial pneumonia (—).

{kind=link}
{kind=link}
Survival in patients with treated (—) and untreated (---) usual interstitial pneumonia.
Univariate analysis for the UIP group, showed that of age, sex, smoking, pulmonary-function tests and treatment with steroids, only carbon monoxide diffusing capacity of the lung (DL,CO) (p=0.01, n=33) and carbon monoxide transfer coefficient (KCO) (p=0.03, n=31) at time of diagnosis predicted survival (low values predicted poorer survival) with steroid usage (p=0.03, n=36) also predictive of poorer survival. By contrast, a multivariate analysis examining all demographic variables, smoking history and pulmonary function, showed that only a history of current or former smoking was found to be a statistically significant predictor of poorer survival (p=0.02).
Discussion
This study of biopsy-proven cases of idiopathic interstitial pneumonias shows some variation from series published from other countries.
Sex distribution in this group of patients with UIP and NSIP showed a preponderance of males in the UIP group but significantly more females presenting with NSIP compared to other studies 1, 2, 6–8, 10, 17, 18. The mean age for this UIP group was significantly higher than that of the NSIP group as has been previously reported, although Daniil et al. 7 had an even lower mean age for a group of 15 NSIP patients at 43 yrs. The duration of symptoms prior to diagnosis between the UIP and NSIP groups in this study did not differ significantly and was longer than in previous series 1, 2, 6, 7 except for the study of Cottin et al. 18 of twelve patients with NSIP who had an average symptom duration of 31±58 months. Further distinctions on clinical grounds between the NSIP and UIP groups in this study were not significant apart from a difference in FEV1% pred and residual volume % pred, the latter potentially confounded by copathology such as emphysema. The authors' findings regarding antinuclear antibodies in these two groups of patients replicate observations documented in previous studies where up to 21% of patients with lone IPF have been found to have ANA in low titre 3, 15, 19. Furthermore, none of the patients in this study had an identifiable connective-tissue disease, either clinically or historically. The difference in survival between the NSIP and UIP groups is significant, suggesting that they may represent two distinct disease entities, distinguishable on biopsy alone. This finding reflects data reported previously. The UIP group in this study further demonstrates the best median survival, to date, of published series. Travis et al. 3 reported a median survival of 4 yrs for their UIP group, Bjoraker et al. 2 and Daniil et al. 7 median survivals of 2.8 yrs and Nicholson et al. 8 a median survival of ∼2.5 yrs. The median survival for the UIP group in this study was 6.5 yrs (78 months), even when controlled for demographical, lung function, and treatment and smoking variables. Furthermore, no significant difference in overall survival between treated and untreated patient groups with UIP was found. Survival time was taken from the date of diagnosis on lung biopsy, as has been the case in previous studies 2, 3, 6–8.
The potential biases that could have affected these results were examined, including that of case selection. There is a potential of referral bias to a tertiary centre for cardiothoracic disorders, but 85% of this UIP group were referred from the surrounding suburbs and towns. This does not differ from the referral pattern reported by Bjoraker et al. 2. All patients were fit enough to undergo biopsy (excluding the two patients diagnosed definitively at autopsy) and in contrast to the study by Utz et al. 20, not even one death postlung biopsy was documented, even after 30 days. Lead-time bias could theoretically have explained the long survivals, but only if patients had had biopsies performed unusually early in the course of their disease. This was definitely not the case. Respiratory function did not differ significantly in these groups of patients from those published in similar case series in the literature 2, 6, 7, although there appears to be a trend in the study by Utz et al. 20 towards a lower DL,CO% pred in patients with UIP. All cases showing biopsy changes of pure honeycomb lung were excluded from this series on the basis that it represents an end-stage process for which the aetiology cannot be determined with precision. The exclusion of such material has not been documented in previous studies 1, 2, 7, 8. The potential for misclassification of histology was minimized as histological diagnosis was achieved via interobserver agreement. Even though four biopsies were obtained by transbronchial biopsy alone, all specimens were considered to show sufficient material to enable accurate diagnosis 21.
All the patients with UIP in this study were Caucasian but drawn from a multi-ethnic general Australian population. It is beyond the scope of this paper to examine the possible role of the environment in affecting survival. Finally, are a different spectrum of patients in whom the natural history of this condition is more benign being seen? This would support the authors' clinical impression that there is a subset of patients in whom the disease remains indolent for prolonged periods of time or fails to progress. This observation also raises implications for potential treatment strategies currently under investigation and the further issues of patient selection and treatment timing.
To conclude, this study highlights differences between populations with interstitial pneumonia, demonstrating a longer median survival in patients with usual interstitial pneumonia with no significant difference in survival between treatment and nontreatment groups. This raises questions concerning the timing of treatment using conventional protocols and supports speculation regarding possible differential genetic susceptibility to the progression of usual interstitial pneumonia.
Acknowledgments
The authors would like to thank P. Lunney for assistance with statistical analysis.
- Received May 10, 2001.
- Accepted December 10, 2001.
- © ERS Journals Ltd
References









