





Abstract
A new study identifies increases in the calcium activated chloride channel, TMEM16A, in the pulmonary arteries of IPAH patients and proposes the repurposing of benzbromarone, a non-specific inhibitor of the channel, as a novel therapy for the disease http://bit.ly/2ITN4ul
One of the most well-established hallmarks of pulmonary arterial hypertension (PAH) is the unique cellular phenotype of the pulmonary arterial smooth muscle cells (PASMCs) from PAH patients. This phenotype, which is defined by hypercontractility, excessive proliferation, apoptosis resistance and aerobic glycolysis, is intimately linked with plasma membrane depolarisation and elevated concentrations of cytosolic calcium ([Ca2+]cyt) [1–3].
Over the past two decades, multiple studies have identified the altered expression or regulation of a variety of cation channels that contribute to this depolarised PASMC phenotype in PAH. These changes include the increased expression of store-operated Ca2+ channels, including the canonical transient receptor potential (TrpC) channel, TrpC6 [4], and a reduction in voltage-gated potassium channels (Kv), such as Kv1.5 [5], which enable the efflux of K+ ions from the intracellular space. Together, increased Ca2+ influx and reduced K+ efflux contribute to membrane depolarisation and a further elevation of [Ca2+]cyt via the opening of voltage-dependent calcium channels (figure 1). More recently, genetic screening of both heritable PAH families and idiopathic PAH (IPAH) patient cohorts enabled the identification of heterozygous loss-of-function mutations in KCNK3, the gene encoding the TASK-1 potassium channel, in both patient populations [6, 7]. KCNK3 is decreased in the monocrotaline rat model of PAH and its inhibition was found to induce aberrant pulmonary vascular cell proliferation in control rats [8]. Moreover, KCNK3 silencing promotes PASMC membrane depolarisation in vitro, offering yet another mediator of the diseased cellular phenotype in PAH [9].

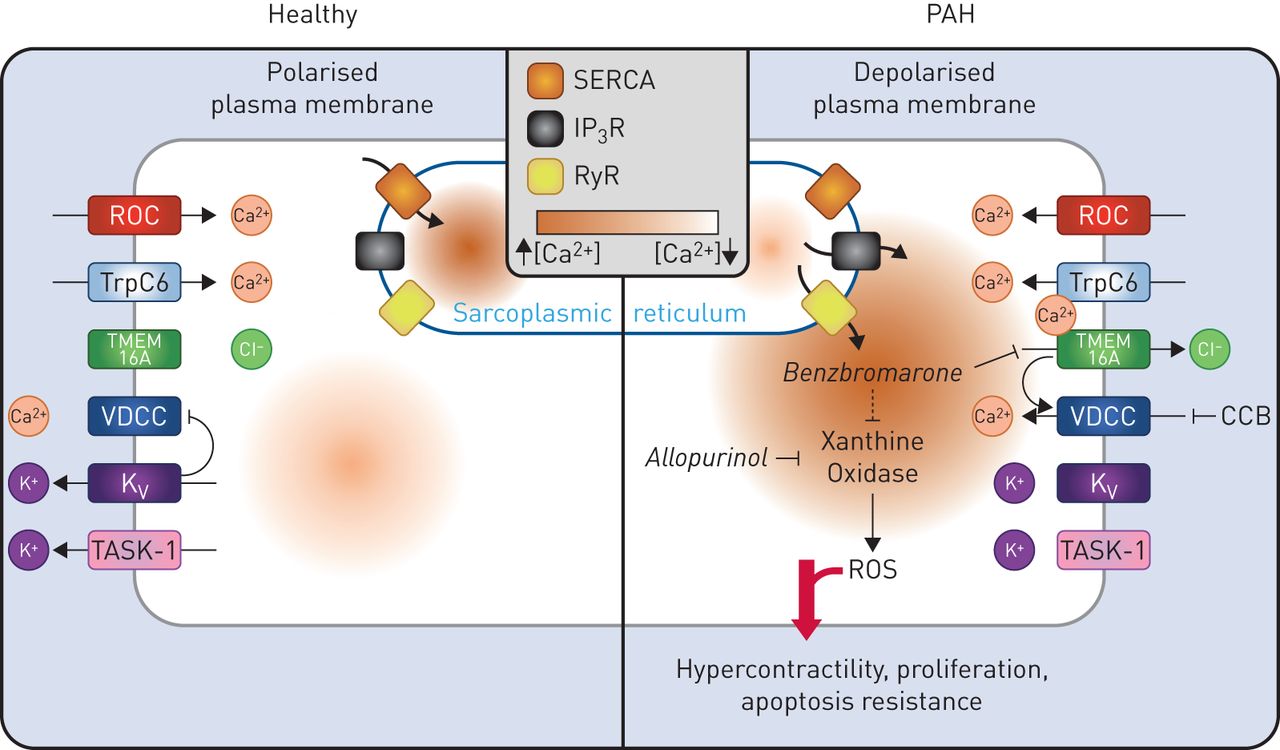{kind=link}
Schematic diagram summarising altered ion channel function in pulmonary arterial smooth muscle cells (PASMCs) from pulmonary arterial hypertension (PAH) patients and the proposed role for benzbromarone in reversing the cellular phenotype of disease. In PAH, store operated calcium channels, such as the canonical transient receptor potential (TrpC) channel TrpC6, are upregulated while potassium channels, including TASK-1 and voltage-gated potassium channels (Kv), are downregulated, leading to increased levels of cytosolic calcium and PASMC membrane depolarisation. These changes drive the activation of voltage-dependent calcium channels (VDCCs). Elevated expression of the Ca2+ activated Cl− channel, transmembrane member 16A (TMEM16A), results in further PASMC membrane depolarisation, hypercontractility, proliferation and apoptosis resistance. Treatment of PASMCs with benzbromarone is believed to block TMEM16A, restoring membrane polarisation and reversing the PAH PASMC phenotype. However, benzbromarone, like allopurinol, may also enact therapeutic benefits through its actions on xanthine oxidase. CCB: calcium channel blockers; IP3R: inositol trisphosphate-sensitive receptor; ROC: receptor operated channel; RyR: ryanodine receptor; ROS: reactive oxygen species; SERCA: sarcoplasmic/endoplasmic reticulum calcium ATPase.
In comparison to this thoroughly defined role for cation handling in PAH, the potential contribution of anion channels to disease has received relatively little attention. To date, this work has largely been limited to studies of the Ca2+ activated Cl− channel, TMEM16A, which is upregulated in the PASMCs of rats exposed to either chronic hypoxia or monocrotaline and serves as a critical contributor to PASMC depolarisation and pulmonary artery hypercontractility in both models of disease [10, 11]. Interestingly, studies in airway epithelial cells have shown that Ca2+ activated Cl− channels, which enable Cl− efflux in response to sub-micromolar increases in [Ca2+]cyt, can function in tandem with TrpC6, providing a possible link between increased TMEM16A activity and a known actor in the depolarisation of PAH PASMCs [12]. Despite this compelling evidence, work supporting an important role for TMEM16A, or other anion channels, in the pathogenesis of PAH has yet to move beyond animal models.
In the current issue of the European Respiratory Journal, Papp et al. [13] address this limitation using a targeted screen of anion channel and transporter gene expression in laser capture microdissected pulmonary arteries from the lungs of IPAH patients and healthy control subjects. The authors identify TMEM16A as the only anion channel that is significantly upregulated in IPAH pulmonary arteries and demonstrate a central role for this channel in PASMC depolarisation. While these studies provide essential insight into the cellular pathobiology of the human condition, much of the value of the work by Papp et al. [13] comes from their introduction of benzbromarone, a non-specific inhibitor of TMEM16A, as a potential PAH therapy.
Benzbromarone is a uricosuric agent and an inhibitor of xanthine oxidase that was first introduced in the 1970s as a treatment for gout [14]. It was not until 2012 that the drug was also identified, through a high-throughput screen, as an inhibitor of TMEM16A at micromolar concentrations [15]. Papp et al. [13] demonstrate that benzbromarone restores membrane polarisation and blocks the platelet-derived growth factor-induced proliferation of PAH PASMCs in vitro, while preventing the development of pulmonary hypertension in vivo in both the chronic hypoxia mouse and the monocrotaline rat model of PAH.
With this study, benzbromarone becomes the latest addition to a long list of existing drugs, including dichloroacetate [16], tacrolimus [17], hydroxychloroquine [18] and others, which are being considered for repurposing as a PAH therapy. As reviewed recently by Prins et al. [19], drug repurposing offers a great advantage for rare and deadly diseases like PAH by allowing for the fast-tracking of regulatory approval, while minimising the costs associated with new drug development. It is noteworthy that benzbromarone is the third gout treatment that has been considered for repurposing in PAH, along with colchicine and allopurinol. Importantly, both allopurinol and benzbromarone are inhibitors of xanthine oxidase, raising the possibility that at least a portion of benzbromarone's therapeutic benefit, beyond any actions on TMEM16A, may be attributable to its impact on the generation of reactive oxygen species (ROS) [20].
When considering the potential repurposing of any drug for use in PAH, it is essential that the candidate demonstrate both safety and efficacy in the human patient population. Regarding the former, benzbromarone may be at a disadvantage relative to other repurposing candidates. Although benzbromarone is still available as a generic drug in several countries, concerns over hepatotoxicity caused Sinofi-Synthélabo to withdraw the drug from the market in 2003. A subsequent risk–benefit analysis suggested that these safety concerns may have been overblown [21]. However, in order to be clinically viable, benzbromarone must still demonstrate sufficient efficacy to outweigh any potential risks to patient safety. In this regard, it is important to acknowledge that benzbromarone was efficacious in preventing the onset of disease in multiple rodent models, but has not yet been shown to reverse established disease in these models, or in the sugen-hypoxia rat model of PAH, which has now become the gold standard pre-clinical model for the translation of potential PAH therapies [17, 22].
Despite the absence of these data, Papp et al. [13] did examine the capacity of benzbromarone to induce an acute vasodilatory response in a small group of 10 PAH patients. In these subjects, the administration of 200 mg benzbromarone, twice the standard daily dose for the treatment of gout, failed to reduce mean pulmonary arterial pressure or pulmonary vascular resistance and actually caused a small but significant increase in both values. Perhaps this result is not surprising when considering previous clinical experiences with L-type, voltage-dependent calcium channel blockers (CCBs). CCBs, such as amlodipine, nifedipine or diltiazem, only induce an acute haemodynamic response in a subset of IPAH patients [23] and are effective for chronic administration in just a small fraction of these individuals, representing the 5–10% of PAH patients for whom excessive and chronic vasoconstriction is a dominant feature of their disease [24]. Although chronic administration of benzbromarone may be required to achieve a beneficial effect in the broader PAH population, it is also possible that targeting altered anion handling may only be a viable therapeutic strategy for a small percentage of PAH patients. If this latter situation is the case, one must also consider whether this subset would represent the same group of individuals that is already clinically well served by CCBs, or if targeting altered anion flux will open up new therapeutic options for a currently undefined set of patients.
Independent of the ultimate clinical value of benzbromarone as a therapeutic strategy, it is worthwhile noting that, with the adoption of CCBs and sildenafil as approved therapies, PAH is already a drug repurposing success story. Moreover, all other approved treatments for PAH originated as designated orphan drugs, dating back to the first use of epoprostenol in 1984 [25]. This history exemplifies the advantages of drug repurposing and expedited drug designations as mechanisms to provide therapeutic options for a relatively small patient group in desperate need of treatment. However, this track record is also a reminder that the field has yet to develop an approved therapy that was custom designed exclusively for PAH, based on the unique molecular pathobiology driving disease progression.
With the rise of high-throughput DNA sequencing technology, a number of new PAH-associated genes have recently been identified to accompany the initial identification of mutations in BMPR2, the gene encoding the bone morphogenetic protein (BMP) type II receptor (BMPR-II), nearly 20 years ago [26–30]. These genes have served to identify novel dysregulated signalling pathways in disease and shed light on the cellular processes that govern the initiation and progression of PAH. More importantly, these new targets also offer the promise of a next generation of tailor-made molecular therapies that are grounded not just in animal models or the cellular phenotype of patients with established PAH, but in the genetics underlying the human condition. Interestingly, a number of these newly identified genes include components of the BMPR-II signalling pathway, such as the BMPR-II ligands BMP9 and BMP10 [28–30], reinforcing the importance of this receptor to disease and supporting the pursuit of therapeutic approaches that aim to restore dysfunctional BMP signalling in PAH. As this work towards the next generation of PAH therapies proceeds, the question remains whether such custom therapeutics are required to truly change the clinical landscape or whether the recycling of drugs that were developed to treat other, unrelated pathologies represents the preferred path forward.
Footnotes
Conflict of interest: A.L. Theilmann has nothing to disclose.
Conflict of interest: M.L. Ormiston has nothing to disclose.
Support statement: This work was supported by the Canadian Institutes of Health Research (grant PJT-152916). Funding information for this article has been deposited with the Crossref Funder Registry.
- Received March 23, 2019.
- Accepted March 28, 2019.
- Copyright ©ERS 2019
References










