





Abstract
The cellular and molecular pathways in asthma are highly complex. Increased understanding can be obtained by unbiased transcriptomic analysis (RNA-Seq). We hypothesised that the transcriptomic profile of whole human endobronchial biopsies differs between asthma patients and controls.
First, we investigated the feasibility of obtaining RNA from whole endobronchial biopsies suitable for RNA-Seq. Secondly, we examined the difference in transcriptomic profiles between asthma and controls. This cross-sectional study compared four steroid-free atopic asthma patients and five healthy nonatopic controls. Total RNA from four biopsies per subject was prepared for RNA-Seq. Comparison of the numbers of reads per gene in asthma and controls was based on the Poisson distribution.
46 genes were differentially expressed between asthma and controls, including pendrin, periostin and BCL2. 10 gene networks were found to be involved in cellular morphology, movement and development.
RNA isolated from whole human endobronchial biopsies is suitable for RNA-Seq, showing different transcriptomic profiles between asthma and controls. Novel and confirmative genes were found to be linked to asthma. These results indicate that biological processes in the airways of asthma patients are regulated differently when compared to controls, which may be relevant for the pathogenesis and treatment of the disease.
Abstract
Transcriptome sequencing shows processes in the airways of asthmatics are differently regulated at transcriptomic level http://ow.ly/kDnln
Introduction
Asthma is associated with a variety of functional changes of the airways including variable airways obstruction [1], increased sensitivity and elevated maximal responses to inhaled irritants [2] and impairment of bronchodilation following deep inspiration [3]. In addition, a process of structural changes called airway remodelling is usually observed in asthma. This includes an increase in airway smooth muscle (ASM) mass, altered deposition of extracellular matrix proteins in- and outside the ASM layer, goblet cell and glandular hyperplasia and angiogenesis [4]. Therefore, the functional and structural characteristics of the airways in asthma are highly complex, which impedes the development of novel targeted therapy [5].
Disease activity in asthma appears to be associated with the pattern and activity of airway inflammation. It has been shown that increased expression of the T-helper cell (Th)2-cytokines interleukin (IL)-4 and IL-13 not only promotes eosinophilic inflammation, but also activates mast cells [6], and that microlocalisation of these mast cells predominantly in the ASM layer is a key factor in the pathophysiology of asthma [7]. Activated mast cells release various mediators, which can lead to enhanced ASM contraction and proliferation [6, 8]. Interestingly, gene expression analysis has shown that the degree of Th2-cytokine expression in bronchial biopsies varies between asthma patients, being associated with aspects of airway remodelling and responsiveness to inhaled steroids [9]. This suggests that there is a close interaction between inflammation and remodelling in asthma.
Airway remodelling is not necessarily a chronic repair response to ongoing inflammation, as traditionally hypothesised. Recent evidence shows that airway remodelling may be driven by pathways that are partly independent of airway inflammation [10]. In addition, airway structural components themselves may drive the pathophysiological process of asthma [11]. Given this complexity of the cellular and molecular pathways, increased understanding of the driving mechanisms in asthma might be best derived from unbiased analysis of bronchial tissue specimens in relation to careful clinical phenotyping [12].
Transcriptomic analysis of the airways by next-generation sequencing (RNA-Seq) allows high-throughput and detailed characterisation of gene expression profiles at the tissue level [13]. Particularly in complex disorders such as asthma, this technology has the potential to discover gene expression profiles that are characteristic of the disease. To our knowledge, there are no head-to-head comparisons of RNA-Seq profiles from bronchial biopsies between asthma and controls.
We hypothesised that the transcriptomic profile of whole endobronchial biopsies is different between patients with asthma and healthy controls. Therefore, the aim of the present study was: 1) to investigate the feasibility of obtaining RNA from whole human endobronchial biopsies suitable for RNA-Seq, and 2) to examine the difference in transcriptomic profiles of whole human endobronchial biopsies between atopic steroid-free mild asthma patients and healthy nonatopic controls.
Methods
Additional information regarding the study methods is included in the online supplementary material.
Design
This cross-sectional study consisted of two visits. At visit 1, subjects were screened for eligibility to participate according to the inclusion and exclusion criteria. This was followed by lung function measurements, including spirometry, forced expiratory volume in 1 s (FEV1) reversibility and a methacholine bronchoprovocation test. At visit 2, a bronchoscopy was performed for the collection of endobronchial biopsies.
Subjects
The study population included steroid-free patients with atopic asthma (n = 4) and healthy nonatopic controls (n = 5). Asthma patients had controlled disease according to Global Initiative for Asthma guidelines [14] and had no exacerbations within the 6 weeks prior to participation. Control subjects had no history or symptoms of pulmonary diseases. This study was part of a larger study registered at the Netherlands Trial Register (the role of the airway smooth muscle layer in asthma; NTR1306).
RNA-Seq and data analysis
Four endobronchial biopsies per subject were collected and frozen after overnight incubation in RNAlater (Qiagen, Venlo, The Netherlands). TRIzol (Invitrogen, Carlsbad, CA, USA) was used to isolate RNA. Amplified cDNA was prepared with the Ovation RNA-Seq System (NuGEN, San Carlos, CA, USA) and cDNA libraries were constructed using SPRIworks Fragment Library System II (Beckman-Coulter, Brea, CA, USA). Emulsion-based clonal amplification was performed using the GS FLX Titanium emPCR Kit Lib-L (Roche, Penzberg, Germany) to prepare enriched DNA library beads for sequencing on the GS FLX+ System (454; Roche). A schematic overview of the strict workflow is presented in figure 1.
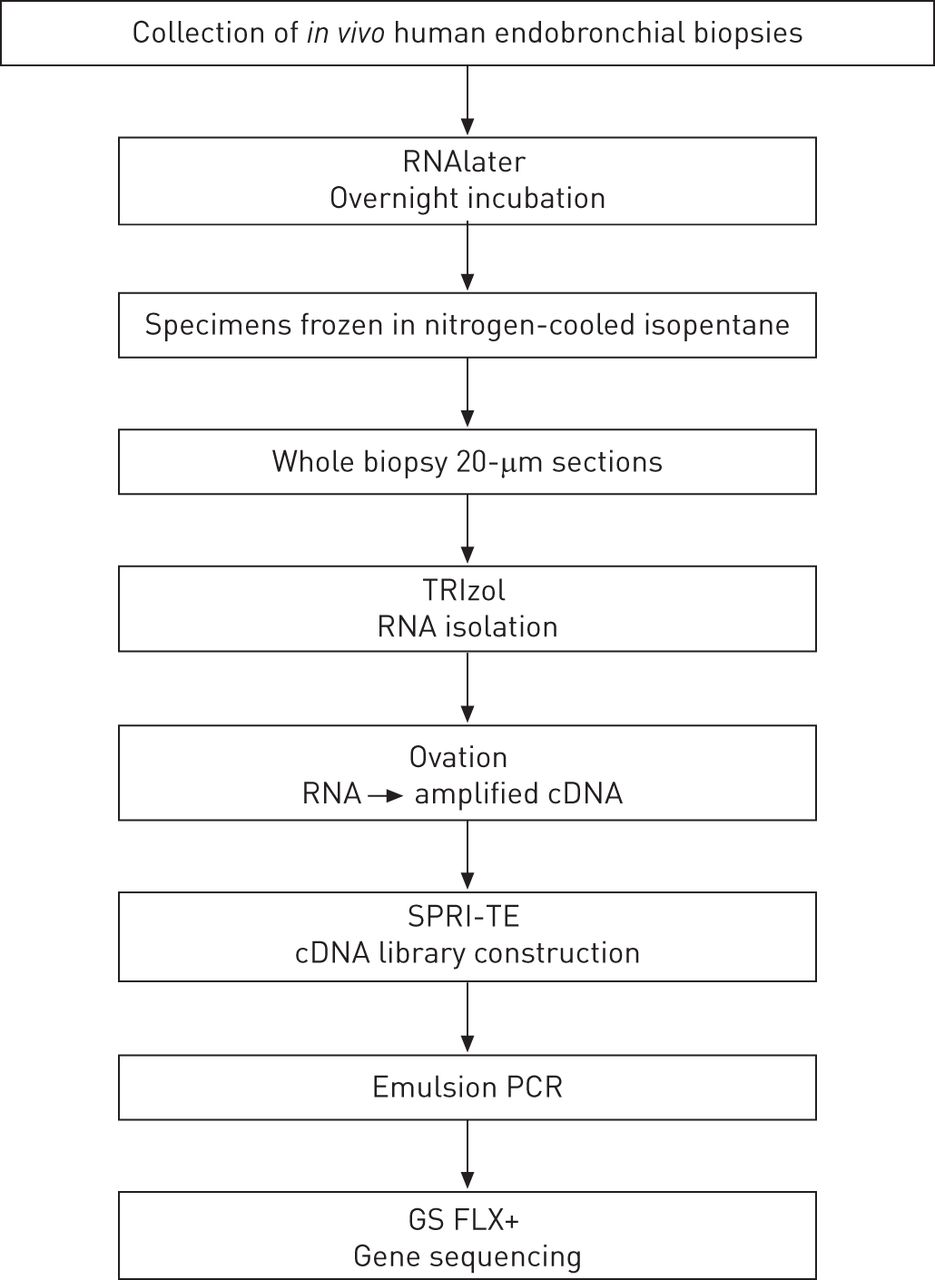
Workflow for biopsy specimens. RNAlater (Qiagen, Venlo, The Netherlands); TRIzol (Invitrogen, Carlsbad, CA, USA); Ovation RNA-Seq System (NuGEN, San Carlos, CA, USA); SPRI-TE nucleic acid extractor (Beckman-Coulter, Brea, CA, USA); GS FLX+ System (Roche, Penzberg, Germany).
Sequencing reads were mapped against the human genome (hg19) [15] using the GS Reference Mapper (Roche). Ingenuity Pathway Analysis (IPA; Ingenuity Systems Inc., Redwood City, CA, USA) was used to identify gene networks. IPA generated a network score, which gives the likelihood that the set of genes in this network could be explained by chance alone. Networks with a score ≥2 have ≥99% confidence that they are not generated by chance. Quantitative PCR (qPCR) was performed to validate the sequencing data. RNA-Seq data was uploaded to the Gene Expression Omnibus (GSE38994). To validate the sequencing data, qPCR was performed on selected genes that were differentially expressed between asthma and controls.
Statistical analysis
Subject characteristics of the study groups were compared using unpaired t-tests or Mann–Whitney U-tests. Comparison of the numbers of reads per gene between asthma and controls was based on the Poisson distribution with correction for total number of reads. Cohen's effect size was calculated to quantify the size of the difference in read numbers of individual genes between asthma and controls. Bonferroni correction for multiple testing was applied. A p-value of <0.05 was considered statistically significant.
Sample size and power calculation
We conservatively assumed that all pathways that are associated with asthma consist of ≥10 000 genes [16], which represents approximately a third of the 31 227 genes of the human genome (hg19) tested. The average fold change of the gene expressions of asthma versus controls was presumed to be about 1.2, which coincides with a Cohen's effect size of about 0.5. A significance level of 0.0001 was used in the statistical analysis to correct for multiple testing. With this significance level and nine subjects in total with four biopsy specimens per subject, we expected to find 48 genes that are truly associated with asthma and two falsely significant genes [17, 18]. This resulted in a false discovery rate of about 4%.
Results
Subjects
table 1 shows the subject characteristics of the asthma patients and healthy controls. Lung function characteristics were largely comparable between the two groups. As expected, the provocative concentration of methacholine causing a 20% fall in FEV1 was lower in asthma patients compared to controls.
Endobronchial biopsy specimens
Four endobronchial biopsies per subject of ∼1–2 mm3 were successfully collected from all study participants. The amount of total RNA isolated from four endobronchial biopsy specimens per study subject ranged from 900 to 9300 ng.
RNA-Seq
Amplified cDNA was prepared from 50 ng of isolated RNA, and had an average length of 250 bp in amounts ranging from 6120 to 9630 ng. cDNA libraries had an average length of 700 bp. The amount of cDNA molecules ranged from 0.25 to 1.58×109 molecules·μL−1. DNA beads with 16–24% enrichment after clonal amplification of the cDNA libraries using emulsion PCR were used for sequencing.
The median read length obtained with the sequencing run was 199 nucleotides with a maximum of 867 nucleotides. Total read numbers of 349 239 and 399 999 were obtained for asthma and controls, respectively. For asthma patients 87% of the reads could be mapped to the human genome hg19, while this was 88% for controls.
The transcriptomes mapped to 10 167 unique genes for asthma and 11 006 for controls (fig. 2). With a medium contig read length of 261 bp, the coverage in the current study is approximately 24× [19]. After correction for multiple testing, 46 genes with an effect size of 0.5 were differentially expressed between asthma and controls (table 2). No significant differences in reference genes were found, including glyceraldehyde 3-phosphate dehydrogenase (GAPDH), β-actin (ACTB), β-2-microglobulin (B2M) and lactate dehydrogenase (LDHA).

Volcano-plot of genes of asthma patients versus controls. The logarithms of the fold changes of individual genes (x-axis) are plotted against the negative logarithm of their p-value to base 10 (y-axis). Positive log2 (fold change) values represent upregulation in asthma compared to healthy controls, and negative values represent downregulation. Circles above the dotted line represent differentially expressed genes between asthma and controls with p<0.05 after correction for multiple testing. DCP1B: DCP1 decapping enzyme homologue B (Saccharomyces cerevisiae); DDB2: damage-specific DNA-binding protein 2 (48 kDa); DYX1C1: dyslexia susceptibility 1 candidate 1; EFCAB3: EF-hand calcium binding domain 3; FKTN: fukutin; POSTN: periostin; SLC26A4: pendrin.
Gene network identification
The 46 differentially expressed genes between asthma and controls were used to identify gene networks by the IPA software application. 10 gene networks were identified with a network score of ≥2. The highest scoring network had a network score of 32 and was associated with the network functions cellular movement, cell death and cellular morphology. The second and third ranking networks, scoring 21 and 17, were associated with genetic disorder and cellular movement and development, respectively (fig. 3). The gene B-cell CLL/lymphoma 2 (BCL2), which was one of the 46 differentially expressed genes between asthma and controls, appeared to be a key component in the network with the highest network score (fig. 3a). Other key components in this network were mitogen-activated protein kinase (MAPK) 1, also known as extracellular signal-regulated kinase (ERK) 1/2, nuclear factor (NF)-κB, p38 MAPK, and transforming growth factor (TGF)-β. 14 of the 46 input genes could be located in this network. The second ranking network included signal transducer and activator of transcription (STAT) 3 and staufen (STAU2) (fig. 3b), while the third network comprised epidermal growth factor receptor (EGFR) and pendrin (SLC26A4) (fig. 3c).

{kind=link}
{kind=link}
{kind=link}
Top three gene networks by Ingenuity Pathway Analysis (IPA) (Ingenuity Systems, Inc., Redwood City, CA, USA). a) The network with the highest network score of 32, including B-cell CLL/lymphoma (BCL) 2, mitogen-activated protein kinase (MAPK) 1, nuclear factor (NF)-κB, p38 MAPK and transforming growth factor (TGF)-β, was associated with the network functions cellular movement, cell death and cellular morphology. b) The second highest scoring network, including signal transducer and activator of transcription (STAT) 3 and staufen (STAU2), and c) third network, including epidermal growth factor receptor (EGFR) and pendrin (SLC26A4), were associated with genetic disorder and cellular movement and development, respectively. The intensity of red- or green-coloured shapes indicates the degree of up- or downregulation, respectively. KB genes: genes included in the IPA Knowledge Base.
Validation of sequencing data
Significant correlations were found between the number of sequence reads per gene and Cq-ratio for tryptophanyl-tRNA synthetase (WARS) (r = -0.652; p = 0.05), Secretoglobin (SCGB) 1A1 (r = -0.671; p = 0.05) and BCL2 (r = 0.724; p = 0.04). These results show that a high sequencing read number corresponded with a low Cq-ratio (high amount of cDNA) and vice versa. Therefore, the qPCR results confirmed the data found by RNA-Seq.
Discussion
In this study RNA isolated from whole human endobronchial biopsy specimens was successfully sequenced, showing transcriptomic profiles of bronchial tissue that were different between asthma and controls. Therefore, RNA isolated from endobronchial biopsies is suitable for RNA-Seq when processed according to a strict workflow. Analysis of the RNA-Seq data revealed expression of novel genes that have not been linked to asthma and airway inflammation previously. Additionally, gene network identification showed that several of the 46 differentially expressed genes between asthma and controls are key components in gene networks associated with the regulation of one or multiple cellular functions, including cell morphology, movement and development. These findings indicate that the regulation of biological processes in the airways is evidently different between asthma and healthy controls, which may be relevant for the pathogenesis and future treatment of the disease.
To our knowledge this is the first study to apply RNA-Seq instead of microarrays to examine the transcriptomic profiles of in vivo human endobronchial biopsies of asthma patients and healthy controls. This high-throughput next-generation sequencing method enables an unbiased way of gene expression analysis, due to the fact that it is not limited to a selection of a number of known genes or sequences, as is the case with microarrays [13]. Therefore, next-generation sequencing facilitates the discovery of de novo transcripts and identification of novel disease-related genes.
We have found 46 genes in bronchial biopsies that were differentially expressed between asthma and controls. Several of these genes have been shown to exert various effects on mRNA degradation and translational processes affecting cellular functions, such as STAU2 [20] and WARS [21]. However, the majority of the differentially expressed genes have not been linked to asthma yet. Previous studies using microarrays have shown that the gene expression profile of airway epithelial cells is different in asthma as compared to healthy controls [22, 23]. This included SLC26A4, also known as pendrin, which in our study was also found to be one of the most prominently upregulated genes in asthma. It has been shown that this gene is closely associated with the pathophysiology of asthma [24]. More specifically, stimulation of the IL4/IL13 receptor complex leads to an increased expression of SLC26A4 via the transcription factor STAT6, resulting in increased mucus production and viscosity of the airway surface fluid [25]. In addition, it has been shown previously that periostin (POSTN), chloride channel regulator 1 (CLCA1) and serpin peptidase inhibitor, clade B, member 2 (SERPINB2) were highly induced in airway epithelial cells [9, 22]. Our current study extends these findings, showing that the expression of these three genes was higher in asthma compared to controls, although only periostin reached significance (table 2). This could be due to the fact that we sequenced the transcriptome of whole bronchial biopsies instead of airway epithelial cells only.
Additionally, input of the 46 differentially expressed genes into the IPA database yielded 10 gene networks that were related to regulatory functions on the cellular level. Besides the fact that several of the 46 genes take key positions in these networks, it should also be noted that particular cytokines, chemokines and protein complexes that are well-known for their major roles in inflammatory processes, such as TGF-β, interferon-α, p38 MAPK and NF-κB, are part of these same networks [26, 27]. In addition to p38 MAPK and NF-κB, two other major nodes present in the highest IPA scoring gene network are BCL2 and ERK1/2. It has been hypothesised that inhibition of BCL2 leads to an upregulated immune response accompanied with an enhanced release of pro-inflammatory cytokines and enhanced efficacy of necrosis of cells [28]. Our results show a significantly decreased expression of BCL2 in asthma compared to controls. Furthermore, phosphorylation of ERK1/2 has been shown to be increased by RANTES and eotaxin, whereas inhibition of ERK1/2 reduced the level of ASM proliferation induced by chemokines [29]. The second and third highest IPA-scoring gene networks (fig. 3b and c) contain STAT3 and EGFR, among others. STAT3 has been shown to be involved as a transcriptional mechanism for thymic stromal lymphopoietin to induce a pro-inflammatory gene expression in ASM cells [30], whereas blocking EGFR may lead to a reduction in mucus hypersecretion [31]. Taken together, these results indicate that the regulation of biological processes in the airways is evidently different in asthma compared to controls. Furthermore, these three gene networks may give further insight into the axis from pathology and inflammation (p38 MPK, NF-κB, BCL2 and EGFR) to physiology (ERK1/2, and STAT3) of the airways in asthma.
Several methodological issues need to be addressed. The inclusion of well-characterised subjects seems to represent a strength of our study. Only steroid-free asthma patients with controlled disease were included, thereby avoiding the potential bias of steroid usage on the gene expression profile of the airway wall. All asthma patients remained stable and controlled with minimal clinical symptoms during the study period. Although they were allowed to use inhaled short-acting β2-agonists as rescue therapy, none of these patients used any during the study period. Therefore, the gene expression profiles could not be potentially influenced by medication usage.
We designed a strict standard operating procedure for the workup of the endobronchial biopsies. Furthermore, by combining the Ovation RNA-Seq System and GS FLX+ we could maximise the utility of the potentially low yield of and even partially degraded RNA from in vivo obtained human endobronchial biopsies. First, for the successful processing of RNA into amplified cDNA, the required input RNA for the Ovation RNA-Seq System is ≥500 pg, which was certainly appropriate for the currently obtained amount of isolated RNA (900–9300 ng). Second, amplification is initiated both at the 3′-end and randomly throughout the transcriptome, thereby maximising the coverage as reads are distributed throughout the whole transcriptome, as well as minimising the potentially detrimental effects of degraded RNA on gene expression analysis. Finally, our sample size was based on detailed power calculations showing an acceptable false discovery rate of 4%.
Gene network analyses by IPA are based on known pathways. The differentially expressed genes found in the current study may be part of novel pathways that are not yet included in the IPA database, even though it is updated weekly with findings from other databases including Entrez Gene, RefSeq, OMIM and Gene Ontology. We used IPA because we aimed to clarify the association of the gene expression profile found in asthma patients with the pathophysiology of asthma. As the field of gene sequencing is advancing our knowledge of various biochemical processes, novel pathways that are associated with asthma may be discovered in future studies.
Nevertheless, there are some limitations to our study. First, the amplification by the Ovation RNA-Seq system could introduce bias due to the generation of short cDNA fragments. This may potentially result in less representative consensus sequences to be found and used for mapping against the reference genome. Consequently, fewer genes than actually present in the samples could be detected. However, it is unlikely that this affected our results, because we generated high coverage depth and the use of the GS FLX+ System ensures longer sequence reads than competing technologies.
Secondly, the biopsy specimens were taken from levels 4–6 of the right lung. As the degree of airway wall remodelling and inflammation in asthma can vary with location in the tracheobronchial tree and with asthma severity [32], it is possible that gene expression profiles of the peripheral airways and in patients with more severe asthma could turn out to be different from those observed in the present study. Subsequent studies focusing on these locations of the airways in asthma patients with more severe disease are needed to complement our current data in order to provide a more comprehensive overview of the gene expression of the asthmatic airways. In addition, temporal variability should be examined, which will be restricted by ethical constraints.
Thirdly, RNA was isolated from whole endobronchial biopsies. We chose to perform whole biopsy analysis, because the contribution of the individual components of the airway wall to the pathophysiology of asthma is still largely unknown [33]. Furthermore, the 46 differentially expressed genes between asthma and controls did not include transcripts of specific inflammatory cells but of various cellular processes instead, indicating that RNA-Seq of whole biopsies was not affected by a difference in inflammatory cells present in the biopsy material.
Based on the currently observed differences in gene expression profiles of whole biopsies, repeating the analysis in selected individual airway components, e.g. epithelium and ASM, by laser capture microdissection seems warranted. This will allow linking gene expression profiles of individual airway wall components to airway function and disease activity. High-throughput next-generation sequencing has the potential to achieve this aim. It could not only lead to the discovery of novel genes or pathways that are vital to the pathogenesis of asthma, but may also qualify as a tool for subphenotyping patients in relation to clinical outcome or treatment responsiveness. After adequate external validation in newly recruited patients, new therapeutic targets could arise, which may facilitate developing essentially novel asthma therapies. In addition, by implementing a systems biology approach in future asthma studies [12], the entire biochemical process, from translation of DNA to RNA to proteins, to the post-translational modification and regulation of these proteins, will be unravelled through the combination of different “omics” technologies, including genomics, transcriptomics, proteomics and metabolomics.
Conclusion
The current study has shown that RNA isolated from whole human endobronchial biopsies was suitable for RNA-Seq. Transcriptomic analysis of whole biopsies revealed 46 genes that were differentially expressed between asthma and controls, with the majority not linked to asthma previously. Furthermore, pathway analysis showed that several of the 46 differentially expressed genes were key components in gene networks associated with the regulation of one or multiple cellular functions. These results indicate that biological processes in the airways of asthma patients are differently regulated at the transcriptomic level when compared to healthy controls.
Acknowledgments
We thank Saheli Chowdhury (Dept of Experimental Immunology, Academic Medical Centre, Amsterdam, the Netherlands) for the support in designing the primer sets for the quantitative PCRs.
Footnotes
This article has supplementary material available from www.erj.ersjournals.com
Support statement: This study was supported by a research grant from the Netherlands Asthma Foundation (project number 3.2.09.065). Thais Mauad is funded by CNPQ (Brazilian Research Council) (302828/2012-9).
Conflict of interest: Disclosures can be found alongside the online version of this article at www.erj.ersjournals.com
- Received July 24, 2012.
- Accepted December 17, 2012.
- ©ERS 2013
References










