





Abstract
Pulmonary hypertension (PH) is a severe haemodynamic disorder in which the pulmonary artery pressure is persistently elevated, leading to right-sided heart failure and death. Recently, chronic myeloproliferative diseases associated with pulmonary hypertension were included in the group 5 category, corresponding to PH for which the aetiology is unclear and/or multifactorial.
In this review we will describe the distinct forms of PH in the context of the myeloproliferative diseases chronic thromboembolic pulmonary hypertension and pre-capillary PH mimicking pulmonary arterial hypertension. The epidemiology, mechanisms and treatment approaches will be discussed.
- Chronic thromboembolic pulmonary hypertension
- essential thrombocytosis
- myeloproliferative disorders
- polycythaemia vera
- pulmonary hypertension
- pulmonary veno-occlusive disease
Pulmonary hypertension (PH) is defined as a group of diseases characterised by an elevated mean pulmonary artery pressure (Ppa) ≥25 mmHg at rest 1–3. The clinical classification of PH comprises apparently heterogeneous conditions which share comparable clinical presentations, pathophysiology and management. The first category of the clinical classification termed “pulmonary arterial hypertension (PAH)” includes idiopathic PAH (IPAH), hereditable PAH and PAH related to risk factors or associated conditions 2, such as connective tissue diseases, congenital heart diseases, portal hypertension, HIV infection and exposure to drugs and toxins such as fenfluramines (table 1⇓). In the 4th World Symposium on PH held in 2008 2, chronic myeloproliferative diseases (CMPD) associated with PH were included in the group 5 category, corresponding to PH for which the aetiology is unclear and/or multifactorial 3. As emphasised in a recent article by Simonneau et al. 3, it is important to better characterise this variant of the disease in order to provide better tools to clinicians who treat these patients with severe comorbidities.
Updated clinical classification of pulmonary arterial hypertension(Dana Point, 2008) 2
The World Health Organization classification of CMPD includes five groups of haematopoietic disorders, some of which are rare or poorly characterised (table 2⇓) 4. The origin of CMPD is a multi-potent haematopoietic progenitor cell with overproduction of one or more of the formed elements of the blood without significant dysplasia. There is a predilection to extramedullary haematopoiesis, myelofibrosis and transformation at varying rates to acute leukaemia. CMPD include chronic myelogenous leukaemia, chronic neutrophilic leukaemia and chronic eosinophilic leukaemia (which primarily express a myeloid phenotype and polycythaemia vera), idiopathic myelofibrosis, and essential thrombocytosis in which erythroid or megakaryocytic hyperplasia predominates. This phenotypic heterogeneity has a genetic basis; chronic myelogenous leukaemia is the result of the balanced translocation between chromosomes 9 and 22 (Philadelphia chromosome) 5, and polycythamia vera, idiopathic myelofibrosis and essential thrombocytosis are characterised by different rates of expression of a JAK2 mutation, V617F 6. This essential distinction is also reflected in the natural history of chronic myelogenous leukaemia, chronic neutrophilic leukaemia and chronic eosinophilic leukaemia, which is usually measured in years, and their high rate of transformation into acute leukaemia. In contrast, polycythaemia vera, idiopathic myelofibrosis and essential thrombocytosis have relatively indolent clinical courses that may be interrupted by recurrent thrombo-haemorrhagic complications. The incidence of CMPD is estimated at between six and nine new cases per 100,000 each year. New cases most commonly occur between the age of 40–60 yrs. Pulmonary complications of CMPD are infrequent and are usually caused either by infection or by venous thromboembolic events. Pulmonary or pleural extramedullary haemopoiesis is a rare complication that may be associated with myelofibrosis 7, 8.
World Health Organization classification of chronic myeloproliferative diseases
EPIDEMIOLOGY
The possible association of PH with CMPD has been suggested by case reports and small case series 9, 10, 11–22. However, the exact incidence and prevalence of PH in this population remains poorly defined. Furthermore, since the clinical signs of PH appear at an advanced stage of the disease and, in some cases, the diagnosis of CMPD is difficult to establish in the context of chronic hypoxaemia, the prevalence of PH is probably underestimated in this patient population. An early study 9 has evaluated cardiac involvement in 30 patients with CMPD (18 females and 12 males). 18 patients had polycythaemia vera, eight essential thrombocytosis and four angiogenic myeloid metaplasia. PH unrelated to valvular disease was identified in four (13%) out of 30 patients. Garypidou et al. 10 have described the prevalence of PH in a cohort of 24 patients with CMPD. Two patients had polycythaemia vera, 14 essential thrombocytosis, six agnogenic myeloid metaplasia and two chronic myelogenous leukaemia. A diagnosis of PH was proposed if the estimated right ventricular systolic pressure (Prvs) was >35 mmHg on Doppler echocardiography. 10 (41.7%) patients, four males and six females, had PH with a mean Prvs of 42 mmHg (37–70 mmHg). None of the parameters analysed in the study which included age, sex, presence of splenomegaly, type, duration and age at diagnosis of CMPD, presence of symptoms, haemoglobin levels, white blood cell or platelet count was predictive of the presence of PH. However, the rate of patients with PH may be overestimated since no gold standard haemodynamic confirmation of PH was obtained and secondary causes, such as left ventricular dysfunction or high cardiac output state, were not excluded 23, 24. Altintas et al. 11 reported on the incidence of PH in 46 patients with essential thrombocytosis. A diagnosis of PH was proposed if a patient had a Prvs >35 mmHg on transthoracic echocardiography. PH was found in 22 (47.8%) out of 46 patients with essential thrombosis and significantly higher platelet counts were observed in patients with essential thrombocytosis and PH. Another study by Gupta et al. 12, which also did not use right heart catherisation (PH was evaluated by Prvs >35 mmHg on transthoracic echocardiography), described PH in 12 (48%) out of 25 patients with CMPD. There was no relationship between PH and age at diagnosis, duration of disease, platelet count and haematocrit level. Since angiogenesis is believed to contribute to the pathogenesis of both PH and CMPD, a recent study has investigated the association of PH and primary myelofibrosis 13. The study group consisted of 36 patients, 22 with primary myelofibrosis, seven with myelofibrosis developing from polycythaemia vera and seven with myelofibrosis progressing from essential thrombocytosis. PH (Prvs >35 mmHg on transthoracic echocardiography) was found in 13 (36%) patients 13.
However, all these studies have major limitations, as the diagnosis of PH was not established as recommended by expert guidelines 23, 24. Therefore, the rate of patients with PH may be overestimated, as elevated Prvs or systolic Ppa might not be confirmed by invasive measures, or could be due to other causes such as left sided heart disease. Indeed Reisner et al. 9 demonstrated that heart disease is common in patients with CMPD, and they reported that 19 out of 30 patients had valvular lesions particularly when their past history was complicated by a thromboembolic event. In addition, some patients may have hypermetabolic state resulting in high cardiac output and passive elevation of the Ppa without elevation in pulmonary vascular resistance. Thus, transthoracic Doppler echocardiography can serve only as a screening tool and a right heart study with full haemodynamic evaluation is mandatory for the correct diagnosis of PH 23, 24.
Two major distinct clinical forms of PH have been described in patients with CMPD; chronic thromboembolic PH (CTEPH) and pre-capillary PH mimicking PAH. Guilpain et al. 14 described six patients with CTEPH (five presented with polycythaemia vera and one with essential thrombocytosis). The diagnosis of CTEPH was simultaneous to that of CMPD in all post-embolic cases, and PH revealed the underlying, untreated myeloproliferative disorder. As a result these patients had significantly higher haematocrit levels than patients with pre-capillary PH mimicking PAH. Conversely, pre-capillary PH mimicking PAH associated with CMPD is diagnosed late in the course of the disease 14–16. In the report by Dingli et al. 15, pre-capillary PH mimicking PAH was diagnosed a median of 8 yrs after recognition of the myeloproliferative disorder (0–26 yrs), while in study by Guilpain 14 pre-capillary PH mimicking PAH occurred later in the evolution of the CMPD (polycythaemia vera in three patients and essential thrombocytosis in one) with a median of 162 months (13.5 yrs) and it was associated with myeloid metaplasia.
CTEPH
Aetiology
The suggested mechanisms for the development of CTEPH are shown in figure 1⇓.
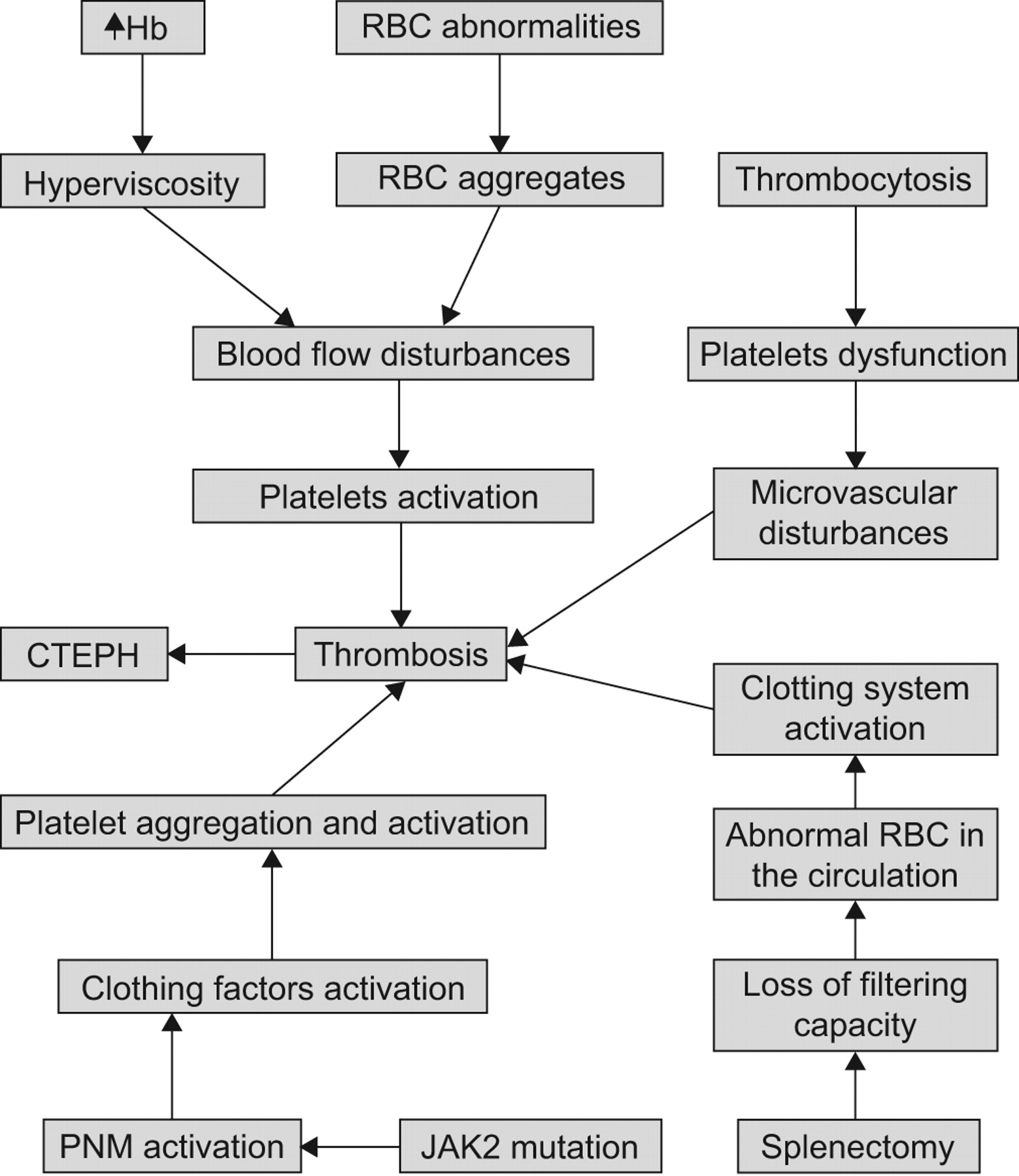
Suggested mechanisms for the development of chronic thromboembolic pulmonary hypertension (CTEPH) in patients with chronic myeloproliferative disorders. Hb: haemoglobin; RBC: red blood cells; PMN: polymorphonuclear cells.
The clinical course of CMPD, and particularly of polycythaemia vera and essential thrombocytosis, is characterised by a thrombophilic state that manifests with microcirculatory disturbances and arterial and venous thromboses 25. These include ischaemic stroke which constitutes 30–40% of all thrombotic events in polycythaemia vera patients, myocardial infarction, peripheral arteries occlusion and deep venous thrombosis of lower limbs, which may cause pulmonary embolism 26. The rate of thrombotic events in polycythaemia vera and essential thrombocytosis is 22–38.6% and 13–29.4%, respectively [21, 22 25–32]. Two thirds of the thrombotic events are arterial and about one third correspond to venous thrombosis in polycythaemia vera, while in essential thrombocytosis most of the thrombotic events are arterial 25–34. In a retrospective analysis of 260 patients with CMPD 27, during a median follow-up of 31 months, thrombotic events were diagnosed in 36% of patients with polycythaemia vera and 20% with essential thrombocytosis. Elevated red blood cell counts, higher haemoglobin levels and increased percentage of nucleated neutrophils at the time of diagnosis and older age were associated with thrombosis. In a retrospective study of the Gruppo Italiano Studio Policitemia 28, patients with polycythaemia vera were included and had been followed for 20 yrs: 41% of patients had at least one thrombotic event, which occurred either at presentation or before diagnosis in 64% of cases. Thrombotic events were seen more frequently in the 2 yrs preceding the diagnosis and were associated with a higher morbidity and mortality. The incidence of thrombosis during follow-up was 3.4% per year. Older patients or those with a history of thrombosis had a higher risk for thrombosis.
The high haematocrit values in polycythaemia vera results in blood hyperviscosity 35. In the venous circulation, characterised by a low shear rate state, hyperviscosity may cause a major disturbance to blood flow and increased risk of thrombosis. At higher shear rates in the arterial circulation the rise in red cell mass displaces the platelets towards the vessel wall, increasing platelet activation and platelet–platelet interactions 21. Furthermore, red blood cell abnormalities which occur in polycythaemia vera and essential thrombocytosis may lead to the formation of red blood cell aggregates that directly contribute to flow disturbance and thrombosis and platelet activation, especially in small vessels 25, 36, 37.
Erythropoietin-independent erythroid colony (EEC) formation is used as one of the criterion for the diagnosis of polycythaemia, although spontaneous EEC formation has insufficient diagnostic specificity and sensitivity as isolated parameters to differentiate between polycythaemia vera and secondary erythrocytosis 4. Spontaneous EEC formation may be associated with thrombosis, particularly of hepatic veins, in the absence of other peripheral blood abnormalities and the presence of EEC leads to the diagnosis of primary myeloproliferative disorder in 78% of cases with apparent idiopathic Budd-Chiari syndrome, and in about half of patients with portal, splenic and/or mesenteric vein thrombosis 38, 39. Another study did not find EEC as a surrogate marker of thrombotic risk (not specifically related to the splanchnic vessels) in male subjects with essential thrombocytosis 40. In the study of Guilpain et al. 14 the diagnosis of CTEPH (fig. 2⇓) and CMPD was made simultaneously in all patients, suggesting that CTEPH could be the first manifestation of CMPD and EEC formation tests might be of interest in order to unmask as yet unrecognised CMPD in CTEPH patients.

Pulmonary angiogram in a patient with polycythaemia vera showing a) the right and b) the left lateral view. The lateral views show evidence of chronic thromboembolic pulmonary hypertension. The patient has been successfully treated with pulmonary endarterectomy. The arrow indicates a pouching defect.
It has been suggested that thrombocytosis can contribute to the vascular events of essential thrombocytosis or polycythaemia vera, since platelet count reduction was reported to lower the risk of microcirculatory disturbances and the antithrombotic efficacy of chemotherapy in high-risk essential thrombocytosis subjects has been clearly demonstrated 25, 32, 41, 42. Essential thrombocytosis patients with microvascular disturbances have decreased platelet survival and evidence for platelet-mediated thrombotic processes, such as increased urinary thromboxane B2 21. Inhibition of platelet cyclooxygenase-1 by aspirin is followed by improving microvascular disturbances with correction of shortened platelet survival time and reduction of thromboxane B2 excretion to normal levels [25 41, 43]. In polycythaemia vera associated with thrombocythaemia, hyperviscosity aggravates the platelet-mediated microvascular disturbances of thrombocythaemia and may cause major arterial and venous thrombotic complications. Correction of hyperviscosity by phlebotomy fails to prevent platelet-mediated microvascular alteration because thrombocythaemia persists 25, 44. However, no correlation was demonstrated in other studies between the degree of thrombocytosis, the presence of platelet dysfunction and the risk of thrombosis in polycythaemia vera 25, 42 In addition, in essential thrombocytosis the correlation with the platelet count appears to be unclear and the antithrombotic effects of chemotherapy may be related to an effect on leukocyte count and, possibly, on platelet and leukocyte activation. Furthermore, marked thrombocytosis can favour haemorrhagic rather than thrombotic manifestations in essential thrombocytosis patients 25, 42, 45.
The increase in white blood cell count is a risk factor for thrombosis in patients with polycythaemia vera and essential thrombocytosis. Previous studies 25, 31, 46 demonstrated an ongoing state of polymorphonuclear leukocytes (PMN) activation, which correlates with increased plasma level markers of clotting activation. PMN activation and increased adhesion of PMN to endothelial cells and platelets support the hypothesis of an involvement of PMN in the pathogenesis of the hypercoagulable state of patients with essential thrombocytosis and polycythaemia vera.
Recently, the presence of the acquired gain-of-function V617F mutation in the tyrosine kinase JAK-2 gene has been demonstrated in PMN and platelets of patients with CMPD 46, 47. The frequency of the JAK2 mutation is close to 95% and 50% in polycythaemia vera and essential thrombocytosis, respectively 47–49. In addition, essential thrombocytosis patients harbouring a JAK2 mutation appear to have a “polycythaemia vera-like” phenotype with increased rate of venous thrombosis 50. Several studies reported on the association of JAK2 mutation and thrombosis at uncommon sites and JAK2 mutation may be associated with increased incidence of thrombosis with no relation to the CMPD. Patients with the JAK2 mutation have increased PMN activation, with platelets aggregation and activation. This may contribute to increased susceptibility to thrombotic events in patients with CMPD 25, 51, 52. Diagnostically in patients with splanchnic vein thrombosis the work-up should include a search for the JAK2 mutation 53. However, in patients with thrombosis outside the splanchnic system and no evidence for CMPD, the JAK2 mutation is infrequently detected and, at present, testing for this mutation is not indicated 54.
In 1966, a retrospective study evaluating 80 splenectomised patients suffering from various types of hereditary haemolytic anaemia found that 13% of the splenectomised patients had thrombocytosis 55. A more recent review of the literature reports that thromboembolic complications following splenectomy for haematological diseases occur in up to 10% of patients and may range from portal vein thrombosis to pulmonary embolism and deep vein thrombosis 56–59. In a prospective case–control study of 109 patients with CTEPH, Bonderman et al 60 found that splenectomy was an independent risk factor for CTEPH.
The pathogenesis of thromboembolism after splenectomy is poorly understood. The transient thrombocytosis expected immediately after splenectomy is usually not associated with thrombotic events and patients may develop CTEPH several years after splenectomy when thrombocytosis is no longer present. Furthermore, many patients have post-splenectomy thrombocytosis for many years without developing signs of thromboembolism suggesting that post-splenectomy thrombocytosis should not be the only factor predisposing patients to thrombosis, although local pulmonary platelets activation and thrombin generation may play a role in the pathogenesis of CTEPH 60–63.
One explanation for the increased thrombotic complication after splenectomy may be the loss of the filtering function of the spleen which allows abnormal red blood cells to remain in the peripheral circulation 61, 62. It is well known that anionic phospholipids of the red blood cell membrane, which are localised on the inner membrane leaflet, can activate the coagulation system. Abnormal exposure of these phospholipids (such as phosphatidylserine) would promote activation of the coagulation process by fixation of enzymatic complexes. Indeed it has been shown in patients with thalassaemia that the number of red blood cells with modified phosphatidylserine expression was 20 times higher after splenectomy 64–66.
In patients with CMPD, splenectomy results in worsening extramedullary haematopoiesis with the potential for pulmonary infiltration. In addition, the platelets in CMPD are functionally abnormal and in concert with the marked elevations in platelet counts it may explain the difference in the interval to the development of PH which is much shorter in patients with CMPD 25. Guilpain et al. 14 reported on 10 cases with PH associated with CMPD. In six of the cases the diagnosis of CTEPH was made simultaneously to that of CMPD suggesting that CTEPH could be the first manifestation of myeloproliferative disorder. In their study the platelet count was not different between patients with CTEPH and PAH associated with CMPD’s. Elevated haematocrit was significantly associated with CTEPH compared to the four patients with pre-capillary PH mimicking PAH associated with CMPD suggesting that elevated haematocrit may contribute to the developments of pulmonary artery thrombosis. Interestingly, none of the patients with CTEPH had undergone splenectomy.
Treatment
Treatment of arterial and venous thromboses in polycythaemia vera and essential thrombocytosis patients should not differ from that recommended in the general population 67.
Pulmonary endarterectomy is the treatment of choice in patients with CTEPH 68. In cases which are inaccessible to surgery, medical therapy including diuretics, anticoagulants, and specific PAH therapy should be considered 69–74. To date, no data on specific PAH therapy is available for patients with CTEPH associated with CMPD.
In patients with high thrombotic risk a cytoreductive therapy with hydroxyurea is recommended 75, 76. Previous studies demonstrated that hydroxyurea reduces the risk of thrombosis in such high-risk patients 25. In the PT1 trial 77, the efficacy and safety of hydroxyurea plus aspirin were significantly higher than those of anagrelide plus aspirin with a reduced number of both thromboses and bleedings in this group. The high antithrombotic efficacy of hydroxyurea can be attributed to its effect on leukocyte count and, possibly, on leukocyte activation 78. The effectiveness of antiplatelet agents in reducing the incidence of vascular events was investigated in several studies. In the ECLAP study 79 a significant reduction of the combined risk of cardiovascular death, non-fatal myocardial infarction, non-fatal stroke and major venous thrombosis without a significant increase of the bleeding risk was demonstrated supporting the use of low-dose aspirin in polycythaemia vera subjects having no contraindication. In essential thrombocytosis, the use of aspirin is controversial since the efficacy and safety of low-dose aspirin has been evaluated only in small-size studies and the potential increased risk of bleeding 25, 80.
PRE-CAPILLARY PH MIMICKING PAH
Aetiology
The suggested potential mechanisms for the development of PAH-like disease associated with CMPD are shown in figure 3⇓.

Suggested mechanisms for the development of pulmonary arterial hypertension (PAH)-like disease associated with chronic myeloproliferative disorders. PDGF: platelet derived growth factor; VEGF: vascular endothelial growth factor.
Several causal factors may be implicated in the pathogenesis of PAH in patients with CMPD. Portal hypertension is a cause of PAH and is a well-known complication of myeloid metaplasia with myelofibrosis 81, 82. Ward et al. 83 reported that 17% of patients with myeloid metaplasia and myelofibrosis develop portal hypertension. First, this may explain the coexistence of the conditions in some patients. Dingli et al. 15 reported that two out of 26 patients with PH associated with CMPD had portal hypertension with oesophageal varices and in the study by Guilpain 14 only one out of 10 patients had portal hypertension with oesophageal varices. Secondly, the use of chemotherapy in these conditions may result in pulmonary damage or promote pulmonary veno-occlusive disease, a rare variant of PH. Pulmonary veno-occlusive disease is increasingly recognised as a cause of PH occurring during the course of cytotoxic chemotherapy and haematopoietic stem cell transplantation 84. A recent study 85 reports a case of biopsy-proven pulmonary veno-occlusive disease as a cause of severe PH in a patient suffering from a CMPD (fig. 4⇓). The introduction of anagrelide several weeks before the development of the symptoms may suggest that this drug might be, at least partially, involved in the pathogenesis of the development of pulmonary veno-occlusive disease. A recent report described six patients with CMPD and elevated Ppa on echocardiography 86. The findings of a low resting saturation, low diffusion capacity for carbon monoxide in concert with the presence of centrilobular ground-glass opacities, septal lines and lymph node enlargement on high-resolution computed tomography of the chest (fig. 5⇓) are highly suggestive of a diagnosis of pulmonary veno-occlusive disease 87. Pulmonary veno-occlusive disease was diagnosed late in the course of the disease and the prognosis was poor 88. Dingli et al. 15 reported that seven out of 26 patients with CMPD and elevated Ppa on echocardiography were treated before or after the diagnosis of PH with anagrelide, but the small number of patients precludes any statistical analysis and most of the patients developed PH without any exposure to the drug.

Pulmonary veno-occlusive disease in a patient with myeloproliferative disease. a) Low-power view showing thickening and partial luminal obliteration of a medium-sized vein in an interlobular septum (arrow), and partial fibrous obliteration of the surrounding lung parenchyma (haematoxylin–eosin stain). b) High-power view of the obliterated vein (haematoxylin–eosin stain). c) Elastin stain showing duplication of the elastic lamina, indicative of arteriolisation of the vein (orcein stain). d) Perl's iron stain showing coarse iron deposits in the pulmonary interstitium, as well as in alveolar macrophages. Scale bars = 100 μm. Reproduced from 80 with permission from the publisher.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
High-resolution computed tomography (CT) of the chest in a patient with pulmonary veno-occlusive disease. The CT scan shows mosaic type, ground-glass opacity of both lungs which is associated with interlobular septal thickening. There is a small amount of right-side pleural effusion (b). The central pulmonary veins and left atrium are normal (not shown). The findings are highly suggestive of pulmonary veno-occlusive disease.
Tumour microembolism leading to PH may be associated with various tumours 89. In patients with progressive myeloproliferative syndrome, megakaryocyte embolism of pulmonary vessels may be caused by translocation of megakaryocytes from the bone marrow, spleen or liver to the lungs, finally leading to PH. Several case reports 87–89 describe patients with CMPD and PH, with pathological examination of the lung demonstrating an obstruction of the small vessels by conglomerates of megakaryocytes. Marvin et al. 17 reported a patient with myeloid metaplasia, PH and right heart failure, thrombocytosis and circulating megakaryocytes after splenectomy. The authors postulated that the cause of PH was pulmonary capillary obstruction by megakaryocytes with stasis and secondary microthrombosis. Hibbin et al. 90 reported that myelofibrosis and myeloid metaplasia is associated with increased numbers of circulating megakaryocytes and myeloid progenitor cells which are poorly deformable and are much larger than the diameter of alveolar capillaries. Therefore, these cells may occlude the pulmonary microvasculature and secrete vasoactive cytokines, which may contribute to the development of PH. Recently, a positive correlation between thrombopoetin levels and systolic Ppa was shown. Since patients with CMPD exhibit increased circulating thrombopoietin levels, thrombopoetin may be related to the pathogenesis of PH in this patient population 91.
Non-hepatosplenic extramedullary haematopoiesis is rare and often associated with myelofibrosis and myeloid metaplasia but it can also accompany other disorders such as polycythaemia vera and melodysplastic syndrome. It is preferentially affects the thoracic spinal region but can involve other organs, such as the lung and the pleura 92. Pulmonary myeloid infiltration during the chronic phase of agnogenic myeloid metaplasia and leukaemic infiltration during the acute transformation of the disease may be related to the development of PH. Steensma et al. 93 described four patients with myelofibrosis with myeloid metaplasia who developed severe PH. In two of the patients technetium-99m sulphur colloid scintigraphy demonstrated diffuse pulmonary uptake consistent with extramedullary haematopoiesis. Weinschenker et al. 94 reported the case of a 76-yr-old female with myelofibrosis and PH in whom an open lung biopsy confirmed pulmonary extramedullary haematopoiesis.
Blood cell proliferation in patients with CMPD may have a major role in pathogenesis of PH. Dingly et al. 15 reported a correlation between Prvs and platelet count in patients with myeloid metaplasia and essential thrombocythaemia, and with haemoglobin levels in patients with polycythaemia vera. As mentioned previously, platelets seem to play a central role in the aetiology of PH. Platelet-derived growth factor released from activated platelets is a strong stimulus for smooth muscle hyperplasia 95, 96, and in an animal model of PH the control of the platelet count slows down the development of PH 97. Previous case studies have described an association between platelet counts, pulmonary pressures and symptoms. Marvin and Spellberg 17 reported that cytoreductive therapy reversed PH and right heart failure in a 72-yr-old patient with myeloid metaplasia by decreasing the platelet count. However, in the study by Dingli 15, 12 out of 26 patients were treated with aspirin before PH was diagnosed suggesting that anti-agregant agents, as opposed to cytoreductive therapy, probably have no effect in preventing or reversing PH.
Finally, recent studies have suggested that enhanced angiogenesis in the peripheral blood and bone marrow might be a possible pathogenetic link between CMPD and PH. In this study patients with primary myelofibrosis and PH had higher bone marrow microvessel density and vascular endothelial growth factor levels suggesting the presence of a pro-angiogenic phenomenon 98, 99. Other studies demonstrated that distinctive features of myelofibrosis associated with PH include normal or low circulating CD34 cell count, polyclonal platelets and granulocytes, absence of peripheral blood dacrocytes and the JAK2 1849G>T(V617F) mutation 100, 101.
Recent reports describe reversible PH in patients with chronic myeloid leukaemia treated with dasatinib, after discontinuation of this multi-targeted tyrosine kinase inhibitor 102, 103. Dasatinib inhibits Bcr-Abl, Src, c-kit, and the platelet-derived growth factor (PDGF) receptor 104, Interestingly, PDGF signalling-related cellular proliferation has been suggested as an important contributor to the development and progression of PAH, and PDGF inhibition has been proposed as a possible therapeutic target in PAH 105, 106. Therefore, further studies are needed to better understand the link between dasatinib therapy and PH in these patients.
Treatment
The evidence for the effectiveness in reducing Ppa with cytoreductive therapy and haematological control of the CMPD is anecdotal. Dingli et al. 15 followed 11 of their patients with serial measurements of Ppa over time, and none of them showed a significant improvement despite good CMPD control. However, another case study described PH reversibility with cytoreductive therapy and reduction of platelet counts, or from therapeutic phlebotomies 17. Several case reports suggest that in patients with CMPD and PH and evidence for extramedullar myelocytosis, a treatment trial with whole-lung, low-dose, external beam radiotherapy may be a useful palliative tool 93, 94. There are no data on the role of anticoagulation and anti-agregants in patients with pre-capillary PH associated with CMPD. However, the antithrombotic strategies in patients with polycythaemia vera and essential thrombocytosis are currently tailored on the basis of the estimated vascular risk 25.
The effectiveness of pulmonary vasodilators used for PAH, including endothelin receptor antagonists, prostacyclin analogues and phosphodiesterase-type 5 inhibitors should be studied further.
Prognosis
The prognosis of patients with pre-capillary PH mimicking PAH associated with CMPD is poor 14–16. In the study of Dingli et al. 15, the median survival time was 18 months and death was mainly related to cardiopulmonary failure. Garcia-Manero et al. 16 reported that the interval between the development of dyspnoea (leading to the diagnosis of PH) and death was <7 months in five out of six patients. In patients with CTEPH, pulmonary endarterectomy is the treatment of choice. In the case study by Guilpain et al. 14 six patients had CTEPH, including four patients with proximal disease. Three patients underwent successful endarterectomy and the fourth patient died after surgery. The two remaining patients had distal CTEPH and were treated with specific PAH therapy.
In summary, the exact incidence and prevalence of PH in patients with CMPD is poorly defined. Two distinct clinical forms of PH were described in patients with CMPD, including CTEPH and pre-capillary PH mimicking PAH. The latter cases are usually diagnosed late in the course of the haematologic disease, while CTEPH is usually diagnosed earlier. High haematocrit levels with hyperviscosity, thrombocytosis and splenectomy, among other mechanisms, may contribute to the increased rate of thrombotic events in patients with CMPD, especially polycythaemia vera. PAH-like disease associated with CMPD was found to be related to myeloid metaplasia suggesting that pulmonary myeloid infiltration and pulmonary capillary obstruction by megakaryocytes with stasis and secondary microthrombosis may contribute to the pulmonary vascular disease. Treatment of PH associated with CMPD is not yet established. Anticoagulant drugs should be administered carefully because of the potential risk of haemorrhagic complications. Cytoreductive treatment should be used in association with symptomatic treatment of PH, such as oxygen and diuretics. There are no data on the effectiveness of specific PAH therapies in these patients and randomised control trials are needed. The prognosis of PH associated with CMPD remains poor. However, pulmonary endarterectomy is the treatment of choice in eligible patients with proximal CTEPH.
Statement of interest
A statement of interest for M. Humbert can be found at www.erj.ersjournals.com/misc/statements.dtl
- Received November 6, 2009.
- Accepted January 4, 2010.
- © ERS
References










