





Abstract
Circulating immune cell populations have been shown to contribute to interstitial lung disease (ILD). In this study, we analysed circulating and lung resident monocyte populations, and assessed their phenotype and recruitment from the blood to the lung in ILD. Flow cytometry analysis of blood samples for quantifying circulating monocytes was performed in 105 subjects: 83 with ILD (n=36, n=28 and n=19 for nonspecific interstitial pneumonia, hypersensitivity pneumonitis and connective-tissue disease-associated ILD, respectively), as well as 22 controls. Monocyte localisation and abundance were assessed using immunofluorescence and flow cytometry of lung tissue. Monocyte populations were cultured either alone or with endothelial cells to assess fractalkine-dependent transmigration pattern. We show that circulating classical monocytes (CM) were increased in ILD compared with controls, while nonclassical monocytes (NCM) were decreased. CM abundance correlated inversely with lung function, while NCM abundance correlated positively. Both CCL2 and CX3CL1 concentrations were increased in plasma and lungs of ILD patients. Fractalkine co-localised with ciliated bronchial epithelial cells, thereby creating a chemoattractant gradient towards the lung. Fractalkine enhanced endothelial transmigration of NCM in ILD samples only. Immunofluorescence, as well as flow cytometry, showed an increased presence of NCM in fibrotic niches in ILD lungs. Moreover, NCM in the ILD lungs expressed increased CX3CR1, M2-like and phagocytic markers. Taken together, our data support that in ILD, fractalkine drives the migration of CX3CR1+ NCM to the lungs, thereby perpetuating the local fibrotic process.
Abstract
The compartmental imbalance of fractalkine mediates the migration of nonclassical monocytes into fibrotic lung tissues. Furthermore, nonclassical monocyte-derived cells show a M2-like and phagocytic phenotype in ILD lungs. http://bit.ly/2CMFWex
Introduction
Interstitial lung disease (ILD) comprises >200 parenchymal disorders, characterised by diffuse interstitial abnormalities and inflammation. ILD can occur in idiopathic forms or as a result of known triggers, such as environmental exposures to inhaled toxins, genetic, autoimmune or infectious triggers. Some ILD subtypes, such as nonspecific interstitial pneumonia (NSIP), hypersensitivity pneumonitis (HP), or connective tissue disease (CTD)-associated ILD may develop into a progressive-fibrotic phenotype with extracellular matrix accumulation that causes impaired gas exchange and leads to respiratory failure [1]. To date, the treatment regimen for ILD consists of drugs that suppress an overactive immune system. While recent studies have suggested that nintedanib leads to better preservation of lung function in ILD [2], the therapeutic response relies on the type of diagnosis and disease severity. Irrespective of the underlying cause of ILD, changes in monocyte or macrophage populations can lead to uncontrolled production of cytokines and growth factors that contribute to lung fibrosis [3, 4]. In IPF, absolute and relative numbers of circulating monocytes associate strongly with decreased survival [5].
Human monocytes can be divided into classical (CM), intermediate (IM) and nonclassical monocytes (NCM) [6]. CM are defined as CD14+++CD16+ and CCR2hiCX3CR1low [7]. CM are well investigated, as they account for ∼85% of the total monocyte population in humans [7, 8]. Chemokine receptor type 2 (CCR2) mediates mobilisation of CM out of the bone marrow to the peripheral blood [7, 8]. The NCM phenotype is defined as CD14+CD16++ and CCR2lowCX3CR1hi [7]. In particular, NCM express high levels of CX3C chemokine receptor type 1 (CX3CR1), which has a unique and exclusive ligand: fractalkine (CX3CL1) [9]. CX3CR1 is needed to actively and continuously patrol the luminal side of the vasculature and to remove damaged cells and debris [10].
A recent single-cell RNA sequencing (scRNA-seq) study reported that a transitional monocyte-derived cell that expresses CX3CR1 gives rise to a disease-associated macrophage in lung fibrosis. This CX3CR1+ CD68+ macrophage transitions into an alveolar identity and localises to the fibrotic niche, where it exerts a pro-fibrotic effect by driving fibroblast accumulation and enhancing fibrosis [11]. In liver fibrosis, CX3CR1+ NCM replenish tissue macrophage pools and regulate immune responses to injury and infection via M2 differentiation [12, 13]. In kidney fibrosis, CX3CR1+ NCM cells induce the generation of reactive oxygen species and the production of fibrotic mediators, such as transforming growth factor (TGF)-β1 or collagen-I [14, 15]. The abundances, specific phenotypes or lung recruitment patterns of monocytes are largely unclear. Here, we sought to characterise monocyte abundances and phenotypes in blood and lung tissue of ILD patients of different origins, explore and characterise the functions of the CX3CR1–fractalkine axis in circulating NCM, and their contributions to tissue-resident phagocyte populations in human ILD samples.
Methods
For a detailed description, please refer to the supplementary material. All reagents used here are listed in supplementary table S1.
Subjects
The study was performed in accordance with protocols approved by the Ludwig-Maximilians Universität München ethics review board (Ethikkommission numbers 180-14 and 454-12). All subjects provided informed written consent for the research study and molecular testing. A total of 105 patients were included in this study (table 1).
Clinical demographics
Flow cytometry
Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient (LymphoprepTM; Stemcell Technologies, Vancouver, Canada). Tissue homogenate CD45+ cells were selected by MicroBeads (Miltenyi Biotec, Bergisch Gladbach, Germany). Peripheral and tissue monocytes subsets were detected using flow cytometry.
Methylprednisolone assay
To investigate the glucocorticoid effect on monocyte subsets, and to understand whether glucocorticoids affect mature monocyte phenotype, we performed experiments with isolated monocytes from controls and treated them with different doses of methylprednisolone, at 3 and 24 h.
Protein quantification
CC chemokine ligand 2 (CCL2) and fractalkine (CX3CL1) (Quantikine Kit; R&D Systems, Abingdon, UK) were used to quantify chemokines levels in plasma and tissue homogenate by ELISA.
Immunofluorescence
Tissue tumour-free areas (control), NSIP, HP and CTD-ILD explanted lungs were used to localise and quantify tissue myeloid cells.
Monocyte adhesion assay
Immortalised murine endothelial cells were used to analyse monocyte adhesion. Monocytes were isolated from PBMCs using the Pan Monocyte Isolation Kit followed by CD16 MicroBeads, resulting in a separation of CD14+ and CD16+ monocytes. We performed a competitive assay between CD16+ monocytes from ILD subjects and CD16+ monocytes from control (supplementary figure S1).
Monocyte migration assay
Isolated monocytes were seeded into a transwell to verify whether migration of monocyte subsets respond to different stimuli. Four different conditions were added in the lower chamber. Migration index was calculated after 3 h (supplementary figure S1).
Statistical analysis
Results are presented as box-and-whisker vertical graphs with mean±sd. For normality distribution, the Shapiro–Wilk test was performed. Therefore, three group comparisons were analysed using the Kruskal–Wallis test followed by Dunn's multiple comparison test. Two group comparisons were analysed using the Mann–Whitney test. Associations between variables were established using linear regression and Pearson correlation. GraphPad Prism (version 7.0; GraphPad Software, San Diego, CA, USA) was used for statistical analyses, and significance was defined as p<0.05.
Results
Details of patient characteristics and additional values and statistical differences between groups can be found in supplementary tables S1 and S2.
Dynamic changes of circulating monocytes in ILD
Monocytes were measured and quantified in circulating PBMCs (figure 1a). Classical monocytes, defined as HLA-DR+CD14+++CD16−, were significantly increased in NSIP and HP patients compared with controls (figure 1b). In contrast, the abundance of intermediate monocytes, defined as HLA-DR+CD14+CD16+, were not different comparing NSIP, HP, CTD-ILD and controls (figure 1c). NCM, defined as HLA-DR+CD14+CD16++, were significantly decreased in NSIP, HP and CTD-ILD subjects, compared with controls (figure 1d). Next, we correlated the percentages of circulating monocytes with diffusing lung capacity of the lung for carbon monoxide (DLCO) and forced vital capacity (FVC). The abundance of CM and NCM were negatively and positively significantly correlated with DLCO % predicted, respectively (figure 1e–g). The abundance of CM, IM or NCM was not significantly correlated with FVC % pred (supplementary figure S2).

Dynamic changes of circulating monocytes population in interstitial lung disease (ILD). a) Peripheral blood mononuclear cells were stained with monoclonal antibodies, and each monocyte subset was previously defined by HLA-DR+ cells and gated according to CD14 and CD16 expression: CD14+++CD16− as classical monocytes (CM), CD14+CD16+ as intermediate monocytes (IM) and CD14+CD16++ as nonclassical monocytes (NCM). Isotype was used as a negative control and is represented by grey dots. Flow cytometry analyses show the percentage of b) CM, c) IM and d) NCM from total monocytes. Control (n=22), nonspecific interstitial pneumonia (NSIP) (n=36), hypersensitivity pneumonitis (HP) (n=28), connective tissue disease (CTD)-ILD (n=19). Statistical analysis was performed using one-way ANOVA with nonparametric Kruskal–Wallis test, followed by Dunn's multiple comparison test. e) Percentage of CM correlated with diffusing capacity of the lung for carbon monoxide (DLCO ) % predicted in ILD (n=58); f) percentage of IM correlated with DLCO % pred in ILD (n=58); g) percentage of NCM subsets correlated with DLCO % pred in ILD (n=58). For statistical analysis, p-values were calculated by t-distribution and Pearson correlation. *: p<0.05, **: p<0.01; ***: p<0.001.
To understand whether monocyte subset abundances were associated with treatment, we grouped all NSIP, HP and CTD-ILD as ILD, and analysed treatment effects as follows. 1) naïve (not treated); 2) immunosuppressive treatment including azathioprine, methotrexate, mycophenolic acid and rituximab (Imm); 3) glucocorticoid treatment; and 4) combined immunosuppressive and glucocorticoid treatment (Imm+GC). This analysis revealed that CM were highest in ILD patients treated with glucocorticoid and Imm+GC, compared with controls, and glucocorticoid-treated compared with treatment-naïve patients (figure 2a). IM abundance was not different between any groups (figure 2b). NCM were lowest in ILD patients treated with glucocorticoid and Imm+GC, compared with controls (figure 2c), and in glucocorticoid-treated compared with treatment-naïve patients (figure 2c).

Circulating mature monocyte abundance is associated with interstitial lung disease (ILD) treatment. Peripheral blood mononuclear cells (PBMCs) were stained with monoclonal antibodies, and each monocyte subset was previously defined by HLA-DR+ cells and gated according to CD14 and CD16 expression, CD14+++CD16− as classical monocytes (CM), CD14+CD16+ as intermediate monocytes (IM) and CD14+CD16++ as nonclassical monocytes (NCM). Flow cytometry analyses show the percentage of a) CM, b) IM and c) NCMs from total monocytes. Control (n=22), ILD naïve (n=15), ILD immunosuppressor (Imm) (n=5), ILD glucocorticoid (GC) (n=31), ILD immunosuppressor with glucocorticoid (Imm+GC) (n=32). Statistical analysis was performed using one-way ANOVA with non-parametric Kruskal–Wallis test, followed by Dunn's multiple comparison test. *: p<0.05, **: p<0.01, ***: p<0.001, compared with control or ILD naïve. d) Different concentrations of methylprednisolone were added or not in 100 µL of whole blood and incubated for 3 h. Flow cytometry was performed to quantify percentages of CM, IM and NCM. e) Different concentrations of methylprednisolone were added or not in cell culture medium of freshly isolated monocytes. Flow cytometry was performed to quantify percentages of CM, IM and NCMs. For methylprednisolone experiments, n=3. Statistical analysis was performed using non-parametric two-tailed Mann–Whitney t-test. *: p<0.05 compared with control, **: p<0.01 compared with control, ***: p<0.001 compared with 24 h without methylprednisolone.
Since peripheral monocyte abundance was associated specifically with glucocorticoid therapy, we next examined whether glucocorticoids influenced monocyte subsets using the following two approaches. First, we treated whole blood with methylprednisolone (at concentrations of 10−12 M, 10−9 M, and 10−6 M) and analysed monocyte phenotypes by flow cytometry. After 3 h of treatment, abundance or phenotype of CM, IM or NCM were not altered compared with untreated blood (figure 2e). When we isolated PBMCs and treated those for 24 h with methylprednisolone (at concentrations of 10−12 M, 10−9 M, and 10−6 M), we observed an increased number of intermediate monocytes treated with 10−9 and 10−6 M of methylprednisolone compared with untreated PBMCs (figure 2e).
Expression of canonical kinetic and scavenger receptors in NCMs in ILD
To fully characterise the immunophenotype of these monocyte populations in ILD, we explored the expression of receptor profiles known to distinguish monocyte subsets (CCR2 in classical monocytes, CX3CR1 in NCMs and CD163 scavenger receptor in myeloid lineages) (figure 3). We observed that CCR2 mean fluorescence intensity (MFI) in CM was significantly decreased in ILD subjects compared with controls (figure 3b–e and supplementary figure S2). CX3CR1 was decreased in ILD subjects in CM and NCM compared with controls (figure 3c and f, supplementary figure S2). In contrast, the scavenger receptor CD163 expression was increased in NCM in ILD subjects compared with controls (figures 3d and g, and supplementary figure S2).

Circulating monocytes show decreased CX3CR1 and increased CD163 expression in interstitial lung disease (ILD). Box and whiskers with dot plot diagrams of flow cytometry analysis show the a) mean fluorescence intensity (MFI) of CCR2, CX3CR1 and CD163; b) CCR2+ classical monocytes (CM); c) CX3CR1+ CM; d) CD163+ CM; e) CCR2+ nonclassical monocytes (NCM); f) CX3CR1+ NCM; and g) CD163+ NCM. a), b), e), f) Control (n=20), ILD (n=83). d), g) control (n=8), ILD (n=57). Statistical analysis was performed using non-parametric two-tailed Mann–Whitney t-test. *: p<0.05, **: p<0.01, ***: p<0.001 compared with control.
CX3CL1 increased NCM migration and accumulation in the lung parenchyma of ILD patients
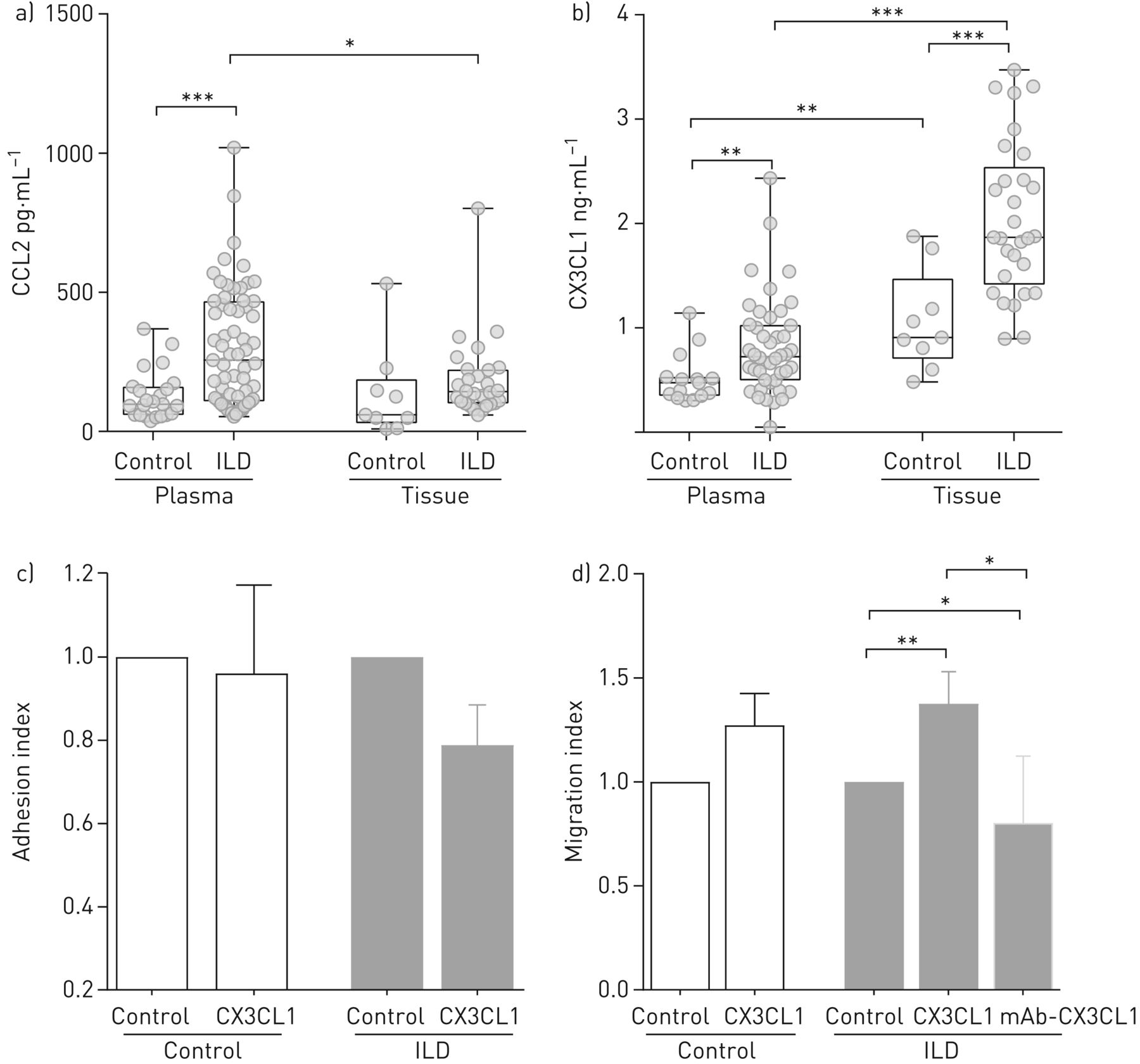
Mobilisation of monocyte populations into and out of the circulation into target organs occurs in a chemokine-controlled fashion [7, 16]. In order to understand the altered abundance of classical and NCMs in the circulation of ILD patients, we investigated chemotactic gradients that might influence peripheral abundance, in particular CCL2 and fractalkine (CX3CL1) in plasma and lung homogenates of controls and ILD. Both CCL2 and CX3CL1 concentrations were significantly increased in plasma of ILD patients compared with controls (figure 4a and b). However, only CX3CL1 concentrations were significantly higher in lung tissues of ILD patients compared with controls (figure 4b). In control and ILD, CX3CL1 levels were higher in the lung tissue than in the plasma.

Fractalkine (CX3CL1) is increased in the lung and drives the migration of nonclassical monocytes (NCM) in interstitial lung disease (ILD). ELISA for CC chemokine ligand (CCL)2 and fractalkine were performed in lung tissue homogenate. a) CCL2 (pg·mL−1) in plasma (control n=23, ILD n=63), and lung tissue (control n=9, ILD n=28); b) CX3CL1 (ng·mL−1) in plasma (control n=15, ILD n=44), and lung tissue (control n=9, ILD n=29); c) in vitro adhesion assay of CD16+ monocytes on the activated endothelial cells (tumour necrosis factor-α, 4 h), control wells were used as an indicator of conversion efficiency (control n=5, ILD n=5); d) in vitro migration assay of NCM, control wells were used as an indicator of conversion efficiency (control n=13, ILD n=14). For functional assays, between three and five experimental replicates were use in each experiment. Statistical analysis was performed using non-parametric two-tailed Mann–Whitney t-test. *: p<0.05, **: p<0.01, ***: p<0.001 compared with control.
Next, we evaluated the effects of this chemoattractant gradient of fractalkine (CX3CL1), and receptor responsiveness to the ligand, in co-cultures of primary monocytes from control and ILD patients with activated endothelial cells. We observed that CX3CL1 did not influence NCM adhesion, but did influence migration (figure 4c). In ILD subjects only, NCM migration was significantly increased in the presence of fractalkine (figure 4d). Blockage of fractalkine (CX3CL1) with a monoclonal antibody reversed this effect (figure 4d). CCL2 had no influence on NCM adhesion or migration (supplementary figure 1).
To characterise localisation of monocytes in the lung, we next performed immunofluorescence staining of lung tissue from ILD subjects and controls. ILD sections identified CD14+CD16+ double-positive cells in lung parenchyma outside vessels (figure 5a). The number of CD14+CD16+ myeloid cells was significantly increased in the lung parenchyma of explanted ILD subjects compared with controls (figure 5a). Immunofluorescence staining showed that CD14+CD16+ double-positive cells expressed CX3CR1 and CD163 in ILD (figure 5b and c), as well as the abundant presence of CX3CR1+CD163+ in ILD lung tissue (figure 5d and supplementary figure S3). CX3CR1+ cells co-expressed the M2 marker CD68 (figure 5e).
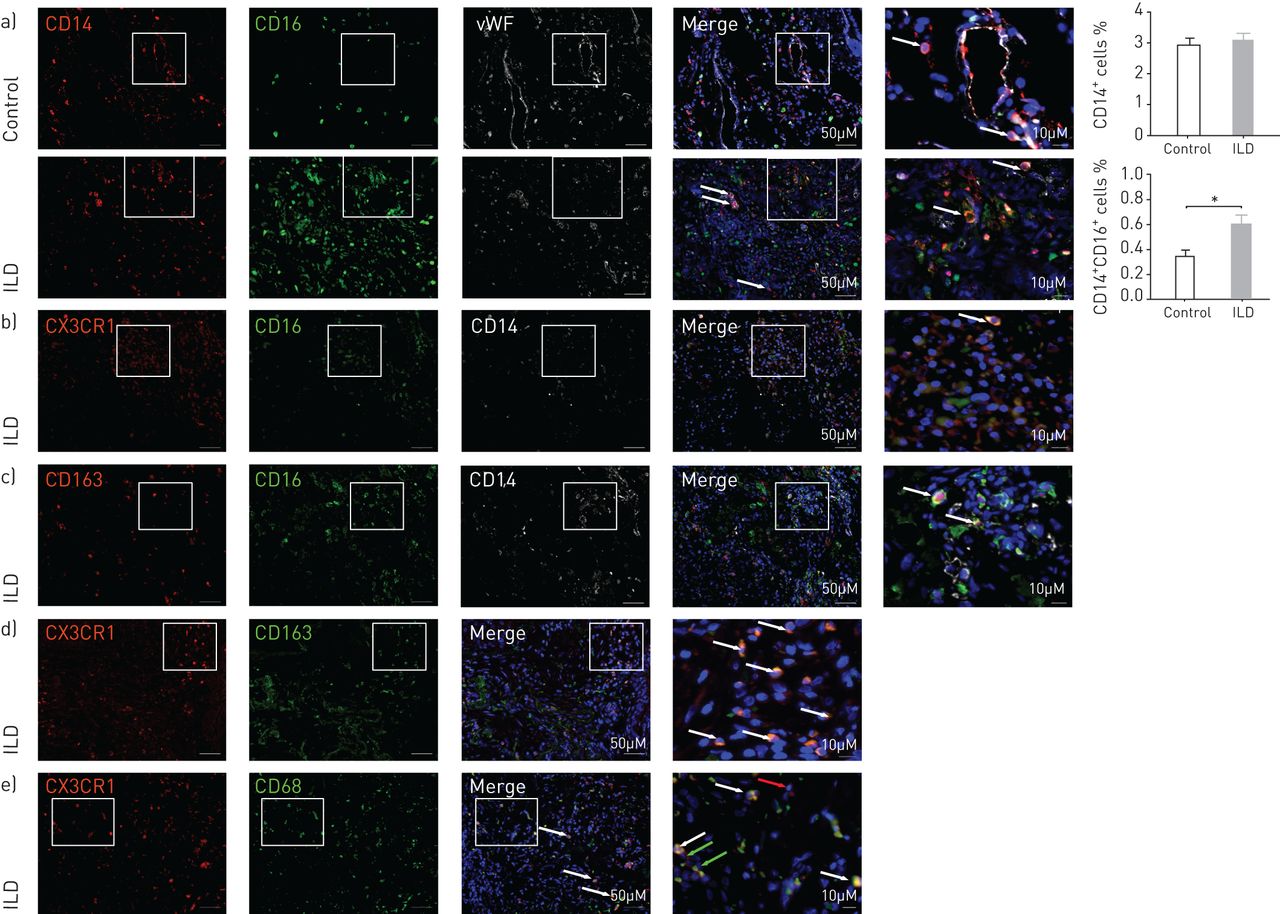
Nonclassical monocytes (NCM) are increased in the lung parenchyma in interstitial lung disease (ILD). Immunofluorescence triple staining was performed using explanted lungs from a) control and ILD. Explanted lungs were stained for CD14 (red), CD16 (green), and von Willebrand factor (vWF) (white). Squares and arrows show double-positive cells; graph bars show the quantification of CD14+ and CD14+CD16+ myeloid cells in the lung parenchyma of control and non-idiopathic pulmonary fibrosis ILD as percentage of total cells. Control (n=3), ILD (n=6). b) CX3CR1 (red), CD16 (green), CD14 (white). Squares and arrows show triple-positive cells. c) CD163 (red), CD16 (green), CD14 (white). Squares and arrows show triple-positive cells. d) CX3CR1 (red), CD163 (green). e) CX3CR1 (red), CD68 (green); red arrows show CX3CR1+ cells and green arrows show CD68+ cells. Squares and arrows show double-positive cells. Cell nuclei are stained with DAPI (blue). All the pictures were taken using magnification of 20× (scale bar=50 µm); white squares represent higher magnification (scale bar=10 µm). Control (n=3), ILD (n=3). Statistical analysis was performed using non-parametric two-tailed Mann–Whitney t-test. *: p<0.05 compared with control.
Increased abundance of pro-fibrotic CD14+CD16++ myeloid cells in the ILD lung
To quantify unequivocally the abundance of CD14+CD16++ double-positive cells in the lung and characterise their phenotype and function, we performed flow cytometry of single cell suspensions of lung tissue. We observed a significant increase in the percentages and absolute cell numbers (cell·µL−1) of CD14+CD16++ double-positive cells in ILD lungs compared with controls (figure 6a). To explore the phenotypic and functional features of tissue NCM, we analysed the expression of mature myeloid markers (CD206) and receptors involved in apoptotic cell clearance and phagocytosis (AXL, MERTK). Flow cytometry analysis showed that tissue NCM in ILD and control expressed CX3CR1 (figure 6b). Tissue NCM from ILD lungs expressed higher levels of MERTK and CD206 than CD14+ cells compared with controls (figure 6b). Immunofluorescence analysis revealed the expression of AXL in CD14+CD16+ cells in ILD (figure 6c). Finally, we performed immunofluorescence staining in ILD lungs to determine which lung cell type contributes to increased fractalkine (CX3CL1) levels. We observed that CX3CL1 was highly expressed in acetylated tubulin-positive airway epithelial cells, but not in collagen-1-positive mesenchymal cells or von Willebrand factor-positive endothelial cells (figure 6d).

Fractalkine (CX3CL1) is expressed by the lung epithelium and nonclassical monocyte (NCM)-derived cells expressed M2-like and phagocytic phenotype in interstitial lung disease (ILD) lungs. a) Flow cytometry analysis and quantification (percentage and absolute numbers) of CD45+ lung cells suspension. Monocytes were gated in CD15− HLADR+ cells (control n=4, ILD n=8); b) flow cytometry staining of CX3CR1, CD206 and MERTK in CD45+ lung cells suspension (control n=4, ILD n=8); c) triple immunofluorescence staining of CD14 (white), CD16 (red) and AXL (green). Pictures were taken using magnification of 40× (scale bar= 20 µm), white squares represent higher magnification (scale bar=10 µm). Cell nuclei are stained by DAPI (blue) (control n=3, ILD n=3); d) immunofluorescence staining of CX3CL1 (red) and acetylated tubulin (Ac. Tub.) or collagen type I (Col1) or von Willebrand factor (vWF) (green) (ILD n=3). CM: classical monocytes.
Discussion
Here, we demonstrate the functional implications of the CX3CR1–fractalkine axis on the abundance and recruitment of monocyte populations in human ILD (figure 7). We demonstrate higher and lower numbers of circulating CM and NCM, respectively, in ILD of multiple different origins. In addition, CM and NCM numbers in peripheral blood correlate with lung function (DLCO) in a negative and positive manner, respectively. These altered abundances of circulating monocytes are the result of predominantly corticosteroid treatment, suggesting that shifting monocyte populations induced by treatment. Functional studies indicate that NCM migration is mediated by fractalkine (CX3CL1), and that ciliated bronchial epithelial cells constitute the local source of fractalkine secretion, creating a chemoattractant gradient of CX3CL1 towards the fibrotic lung. The CD14+CD16++ NCM are abundant in fibrotic ILD lungs and express CX3CR1 and M2-like markers.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
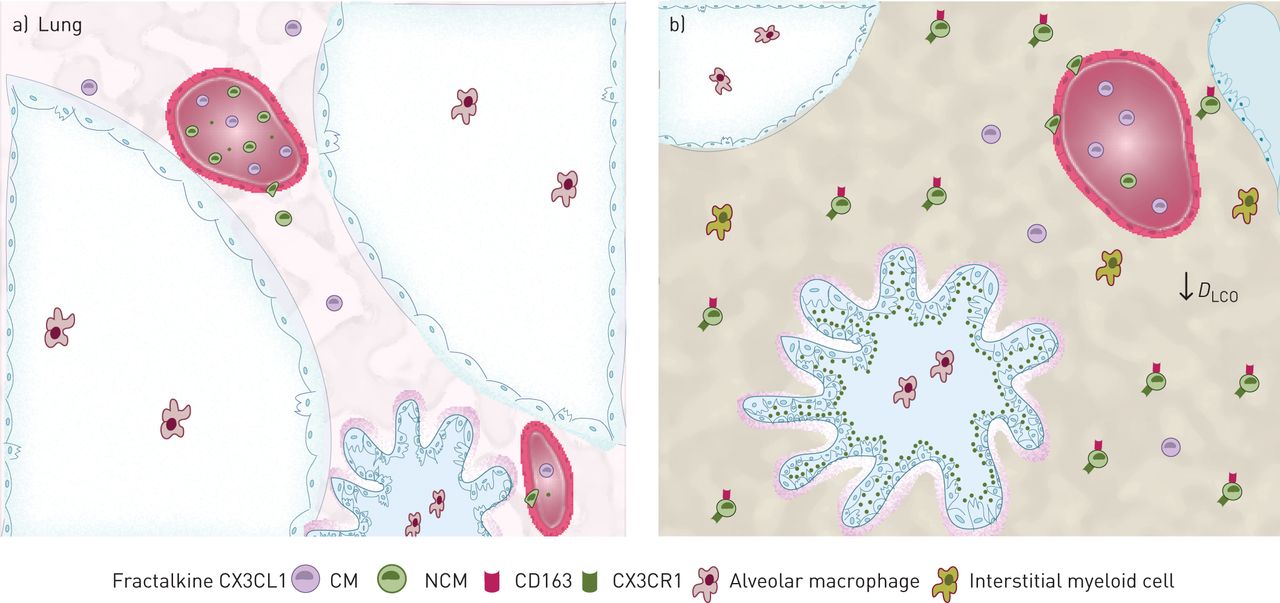
The CX3CR1–fractalkine axis enhances pro-fibrotic nonclassical monocyte (NCM) migration into interstitial lung disease (ILD) lungs. Representative scheme shows an overview of monocyte subsets role and function in a) healthy and b) ILD lungs. NCMs are decreased in the blood of ILD patients. Diffusing lung capacity of the lungs for carbon monoxide (DLCO) is decreased in ILD. Fractalkine (CX3CL1) levels are higher in the lungs than in the blood of ILD patients. Fractalkine is expressed by epithelial cells. CX3CR1+ CD163+ NCM migration is increased in the presence of fractalkine in ILD. CM: classical monocyte.
The percentages of monocyte subsets are highly conserved in healthy humans [7]. However, in acute or chronic diseases, percentages of monocyte subsets may vary and respond differently as a consequence of pathological stimuli [17]. Our results show that CM were significantly increased, whereas NCM were significantly decreased, in the peripheral blood in ILD compared with controls. We observed that patients with higher numbers of CM had worse lung function (as measured by DLCO), while higher numbers of NCM indicated better lung function. Importantly, a recent abstract report of patients from the MESA (Multi-Ethnic Study of Atherosclerosis) study showed that increased abundance of monocytes in the peripheral blood in subclinical ILD is associated with early interstitial lung abnormalities and lower FVC [18], supporting the role of altered monocyte counts even in early stages of disease. However, this study did not report on specific monocyte subtypes. Thus, those data may be driven by the increased abundance of CM in ILD patients, which might occur even at early stages in treatment-naïve patients. More recently, circulating CD14+ classical monocytes have been suggested to be a potent cellular biomarker in idiopathic pulmonary fibrosis (IPF) [5]. Monocytes were quantified in the peripheral blood of IPF patients from six different cohorts showing that high abundance of CD14+ classical monocytes correlated with poor disease outcome [5]. To this end, our cohort is composed of severe and end-stage ILD patients. Concomitant with those previous studies, our data show an increased abundance of classical monocytes in NSIP and HP patients compared with control, as well as a negative correlation with DLCO. Of note, while monocyte counts correlated with DLCO, they did not correlate with FVC, which may be explained by pulmonary vascular changes, differences in clinical handling of IPF and non-IPF ILD, their specific immune subsets or highly dynamic changes of pro-fibrotic monocytes influenced by treatment.
We were interested to understand whether immunomodulatory therapies affected monocyte counts, and thus stratified ILD patients according to treatment regimen. Glucocorticoid therapy has been reported to affect monocyte populations in the peripheral blood [19, 20], and we observed significant effects of glucocorticoid treatment on CM and NCM populations. To further interrogate these effects, we treated whole blood as well as isolated PBMCs, with methylprednisolone in vitro. Interestingly, we observed that glucocorticoid treatment only affected IM, but not CM or NCM abundances, suggesting that the in vivo effects are not due to shifting maturation/differentiation of circulating monocytes, but rather to changes in transmigration patterns into/out of the lung, for example. In line with our experiments, previous studies have described that the abundance of IM is associated with systemic glucocorticoid treatment in uveitis patients [21]. Specifically, the following scenarios may explain the changes of circulating CM and NCM cells in ILD [22]: glucocorticoid-mediated emigration of CD14++CD16− from the bone marrow [23]; a transitory stage of differentiation from CD14++CD16− to CD14+CD16++ [24]; a lack of peripheral maturation of CD14++CD16− cells into CD14+CD16++ cells [25]; or the depletion of CD14+CD16++ monocytes in circulation as they move into tissues [25].
Our phenotypic characterisation of monocytes in the peripheral blood showed that MFI levels of CCR2 and CX3CR1 were decreased in CM, whereas only CX3CR1 was decreased in NCM in ILD patients. When chemokine receptors are engaged in chemotaxis, they can be transiently removed from the cell surface by ligand–receptor internalisation or clathrin-mediated endocytosis [26], which might explain the decrease of CCR2 and CX3CR1 in our data. In contrast, we detected an increased expression of CD163 in NCM in ILD. CD163 is a scavenger receptor and marker of M2 activation [27], expressed by CD14+ circulating cells in scleroderma-ILD patients, and in the tissue of several types of ILD [28, 29].
We observed significantly increased plasma levels of CCL2 and fractalkine (CX3CL1) in ILD. When comparing the circulatory and lung compartments, CCL2 was more abundant in the plasma, suggesting active monocyte recruitment from the bone marrow. This supports the increased numbers of CM in the circulation, and decreased levels of CCR2 by receptor internalisation, as described previously [30]. In contrast, fractalkine levels were higher in lungs than plasma in ILD. Functional experiments demonstrated increased migration of NCM in the presence of fractalkine in ILD samples only. This effect was reversed when a monoclonal antibody against CX3CL1 was added. Monocyte recruitment responds to receptor–ligand interaction [31], our data thus support that NCM migration driven by CX3CL1 is specifically increased during injury. Increasing evidence supports the contributing role of the airway epithelium in diffuse parenchymal lung disease [32–34]. Here we showed that the main source of CX3CL1 in ILD lungs are acetylated-tubulin positive (i.e. ciliated) bronchial epithelial cells. These findings are consistent with a recent study demonstrating that CX3CL1 is expressed by lung epithelial cells in scleroderma-ILD [35] and supports bronchial epithelium-immune crosstalk as an important regulator of tissue injury and fibrosis [36, 37].
In the current study, we performed fluorescence-activated cell sorting analysis of single cell suspensions of lung tissue, and detected increased numbers of tissue NCM in ILD patients. While we are limited in unequivocally determining whether this population constitutes particularly sticky intravascular circulating or true tissue-invading monocytes, similar populations have been recently reported using scRNA-seq data. Providing the first extensive single cell landscape of human fibrotic lungs, Reyfman et al. [37] and Habermann et al. [38] reported a distinct, novel population of pro-fibrotic alveolar macrophages exclusively present in patients with fibrosis. These studies have been extended by the largest scRNA-seq study available to-date using diseased lungs and profiling >300 000 cells from 32 IPF lungs, 18 chronic obstructive pulmonary disease lungs and 29 controls [39]. In this study, the authors detected the presence of pro-fibrotic monocyte-derived macrophages and NCM in fibrotic lungs, although no intra- or extravascular distinction was made. Finally, Travaglini et al. [40] recently provided a human lung cell atlas during fibrosis. By profiling blood and tissue simultaneously, they identified lung-specific immune gene expression profiles and their changes during homing, local signalling interactions, including sources and targets of chemokines in immune cell trafficking. Importantly, they detected the presence of tissue-invading extravascular NCM and their CX3CR1-mediated homing in response to CX3CL1-expressing endothelial cells and airway epithelial cells, confirming our findings [40].
Along these lines, scRNA-seq studies in mice have reported a transitional CX3CR1+ monocyte-derived cell giving rise to fibrotic-associated macrophages in the lung [41]. This CX3CR1+ CD68+ macrophage transitions into an alveolar identity, and localises in the fibrotic niche exerting a local pro-fibrotic effect by driving fibroblast accumulation, myofibroblast differentiation, and enhancing fibrosis [11]. Another complimentary genetic linage tracing study showed that increases in alveolar macrophages during fibrosis are attributable to recruited monocytes-derived cells carrying a fibrotic transcriptome profile [42]. Along this line, our cell culture models using primary lung fibroblasts isolated from fibrotic lungs show increased mRNA expression of pro-fibrotic markers in the presence of CX3CR1+ monocytes and CX3CL1, when compared with CX3CR1+ monocytes alone (data not shown). Furthermore, CX3CR1 knockout mice are protected from fibrosis by decreasing mRNA and protein levels of TGF-β [43].
The CX3CL1/fractalkine is identified as “find-me signal”, from injured pre-apoptotic cells [44]. Therefore, to investigate the functional implication of this finding, we assessed the expression of TAM (TYRO3, AXL and MERTK) family receptors [45] in myeloid cells in the lung. Linked to CD163 scavenger receptor, AXL and MERTK signalling contributes to the “eat-me signals”, which recognise injured and apoptotic cells for phagocytosis by receptor activation [46]. We found that only in ILD, CD14+CD16+ cells expressed AXL. Our results are consistent with previous findings showing that immune cells localised in the fibrotic lung parenchyma are AXL-positive [47]. Furthermore, we show that CD14+CD16++ myeloid cells in the ILD lung expressed CD163, MERTK, macrophage mannose receptor CD206, CD68 and CX3CR1, as supported by others [48].
In conclusion, this study identified a chemotactic gradient of fractalkine that mediates the migration of CX3CR1+ NCM into human fibrotic lungs. Furthermore, NCM-derived cells expressed phagocytosis markers and an M2-like phenotype in the ILD lungs. Targeting the CX3CR1–fractalkine axis may aid the management of end-stage ILD and provide novel therapeutic strategies to either restore monocyte function or modulate recruitment.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-00460-2019.SUPPLEMENT
Shareable PDF
Supplementary Material
This one-page PDF can be shared freely online.
Shareable PDF ERJ-00460-2019.Shareable
Acknowledgements
We gratefully acknowledge the provision of human biomaterial and clinical data from the CPC-M bioArchive and its partners at the Asklepios Biobank Gauting, the Klinikum der Universität München, the Helmholtz Zentrum München and the Ludwig-Maximilians-Universität München. We thank Davide Biondini (Dept of Cardiac Thoracic and Vascular Sciences, University of Padua, Padua, Italy) for fruitful clinical discussions. We thank the CPC Research School (grant writing challenge), the Helmholtz Association, and German Center for Lung Research (DZL) for supporting and funding this work. We acknowledge the support of the European Respiratory Society short-term research fellowship (to F.R. Greiffo).
Footnotes
This article has supplementary material available from erj.ersjournals.com
Author contributions: Conception and design: F.R. Greiffo, O. Eickelberg and I.E. Fernandez. Experimental work, analysis and interpretation: F.R. Greiffo, M. Frankenberger, D. Dietel, V. Viteri-Alvarez, J. Behr, O. Soehnlein, O. Eickelberg and I.E. Fernandez. Intellectual content: F.R. Greiffo, M. Frankenberger, D. Dietel, A. Ortega-Gomez, J.S. Lee, A. Hilgendorff, J. Behr, O. Soehnlein, O. Eickelberg and I.E. Fernandez. Drafting the manuscript: F.R. Greiffo, O. Eickelberg and I.E. Fernandez. Editing the manuscript: F.R. Greiffo, O. Eickelberg and I.E. Fernandez.
Conflict of interest: V. Viteri-Alvarez has nothing to disclose.
Conflict of interest: M. Frankenberger has nothing to disclose.
Conflict of interest: D. Dietel has nothing to disclose.
Conflict of interest: A. Ortega-Gomez has nothing to disclose.
Conflict of interest: J.S. Lee reports grants from NIH, personal fees for advisory board work from Genentech, Celgene and Boehringer Ingelheim, outside the submitted work.
Conflict of interest: A. Hilgendorff has nothing to disclose.
Conflict of interest: J. Behr has nothing to disclose.
Conflict of interest: O. Soehnlein has nothing to disclose.
Conflict of interest: O. Eickelberg reports grants from Ministry of Research and Education Germany, during the conduct of the study; personal fees for advisory board work from Blade Therapeutics, Pieris and Boehringer Ingelheim, outside the submitted work.
Conflict of interest: I.E. Fernandez has nothing to disclose.
Conflict of interest: F.R. Greiffo has nothing to disclose.
Support statement: This work was supported by the Helmholtz Association, German Center for Lung Research (DZL), the CPC Research School, and an ERS short-term fellowship (to F.R. Greiffo). Funding information for this article has been deposited with the Crossref Funder Registry.
- Received June 6, 2018.
- Accepted October 31, 2019.
- Copyright ©ERS 2020
References










