





Abstract
JAK–STAT pathway activation in the lung of COPD patients http://ow.ly/KTwCB
To the Editor:
Chronic obstructive pulmonary disease (COPD) is an inflammatory disease of the lung, most commonly resulting from cigarette smoke exposure, characterised by a largely irreversible and progressive airflow limitation [1, 2]. Currently, there are no disease-modifying therapies to prevent the relentless disease progression in patients with COPD. As a consequence, COPD is associated with high mortality and increasing prevalence worldwide [2]. Therefore, significant research effort has been directed to understanding the mechanism by which cigarette smoke exposure leads to the inflammation seen in COPD in the hope of discovering novel, effective anti-inflammatory therapies to tackle underlying pathogenic mechanisms. Cytokines are thought to be critical orchestrators of the persistent inflammation seen in several inflammatory diseases, including COPD. Many of these cytokines signal through the Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathway and/or are produced as a result of JAK–STAT pathway signalling [3]. Thus, currently, there is considerable interest in developing JAK–STAT inhibitors as a potential treatment for COPD. However, there are no published studies that have comprehensively investigated the activation of all the STAT family members in tissue from COPD patients. Therefore, the aim of our study was to determine the extent of JAK–STAT pathway activation by detecting the presence of phosphorylated STAT proteins 1–6 in lung tissue from nonsmokers, healthy smokers and COPD patients (from transplant surgery) by Western blotting and immunohistochemistry (IHC). The hope was that this would provide important information regarding the role of the JAK–STAT pathway in COPD, which may in turn be useful in the development of novel treatments for COPD.
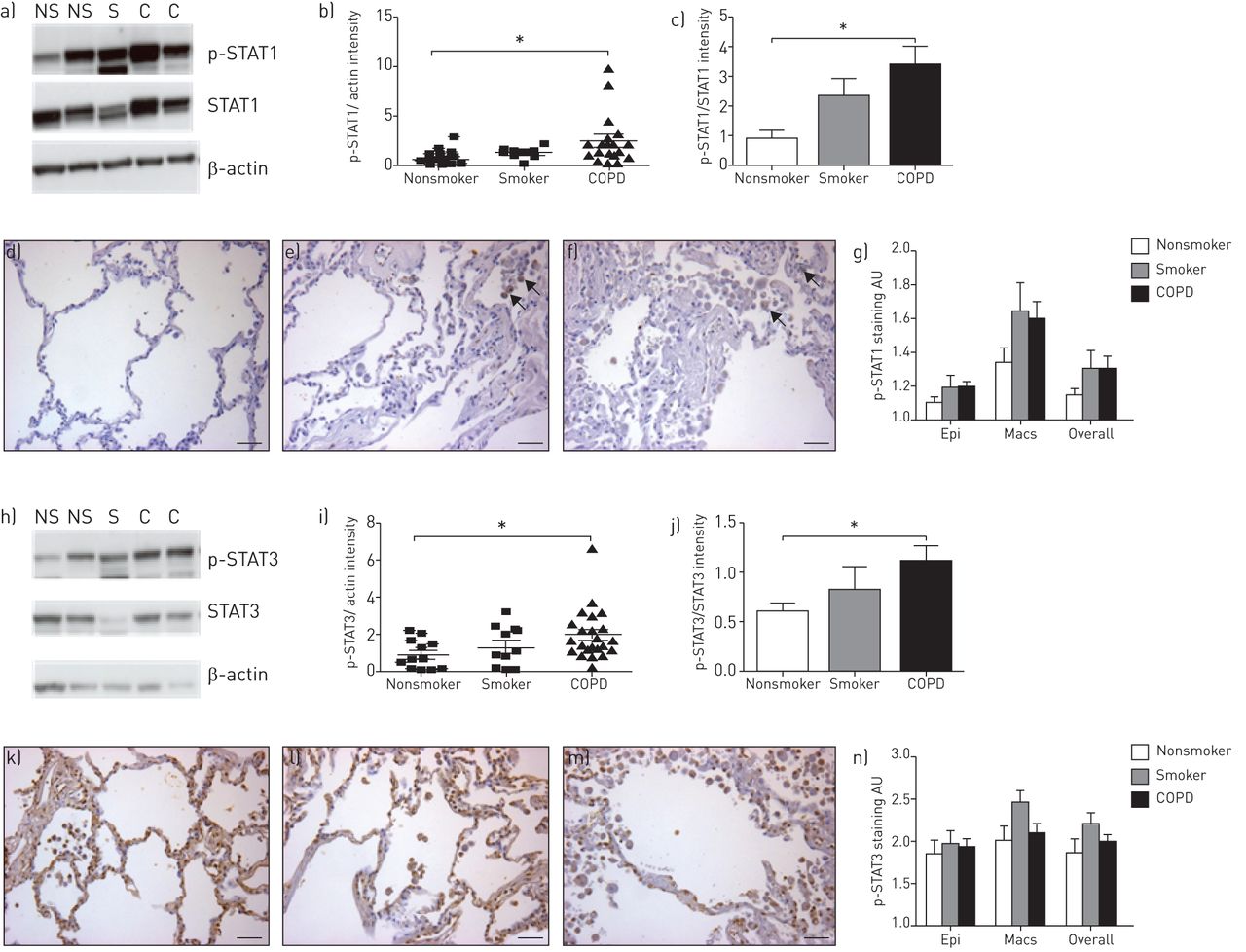
In these studies, Western blotting revealed that tyrosine phosphorylation of STAT1 and STAT3 appeared to be increased in smokers and severe COPD patients compared with nonsmokers, although this only reached significance in the severe COPD patients, indicating that the JAK–STAT1/3 pathway is activated in COPD patients (fig. 1). We could not detect any difference in levels of phosphorylation of either STAT1 or STAT3 at Ser727, or any tyrosine phosphorylation of STAT2, STAT4 or STAT5 in any of the groups. Although we could measure tyrosine phosphorylation in STAT6 by Western blotting, there was no difference between the groups.
{kind=link}
Signal transducer and activator of transcription (STAT)1/3 phosphorylation in human parenchymal samples from nonsmokers, smokers and chronic obstructive pulmonary disease (COPD) patients by Western blotting and immunohistochemistry (IHC). Phosphorylated STAT (p-STAT) proteins 1 and 3 were measured in lung tissue from nonsmokers (NS) (n=12; mean±se 50.1±13.5 years of age; three males and nine females), smokers (S) (n=10; 39.8±13.1 years of age; seven males and three females; no apparent lung disease; the average pack history for the smoking donor group was median (range) 27.8 (14–37) years) and COPD patients (C) (n=23; 55.3±4.6 years of age; seven males and 16 females; lung function data was available for the majority of the COPD patients (n=16): forced expiratory volume in 1 s of 0.85±0.19 L or 30.43±7.13% predicted, forced vital capacity 2.20±0.26 L; all subjects were Global Initiative for Chronic Obstructive Lung Disease stage III/IV; none were active smokers at the time of the lung transplant but previous average pack history was 47.6 (30–80) years) by Western blotting. Protein was extracted from frozen human lung parenchymal samples in the presence of protease and phosphatase inhibitors, and standard Western blotting techniques were used for the investigation of STAT phosphorylation. Primary antibodies were purchased from New England Biolabs (Hitchin, UK) and used at a 1/500 dilution (p-STAT1, p-STAT3, STAT1 and STAT3) except for the β-actin antibody, which was purchased from Sigma-Aldrich Ltd (Poole, UK) and used at a 1/1000 dilution. Example western blots for a) p-STAT1 and h) p-STAT3 are shown. Mean±sem densitometry readings expressed as b, c) p-STAT1 and i, j) p-STAT3 normalised to the b, i) actin loading or c, j) total STAT signal. Standard IHC techniques were used for the investigation of STAT phosphorylation in formalin preserved lung tissue. Primary antibodies for IHC were purchased from New England Biolabs (p-STAT1 and p-STAT3). A donkey anti-rabbit secondary antibody was purchased from Jackson Immunoresearch (Newmarket, UK) and used at a 1/200 dilution. The intensity of the p-STAT staining in each sample was scored by eye using a light microscope. Two independent observers, blinded to the sample group to avoid bias, scored the slides. For each slide, the staining intensity was graded relatively as 1–4 from no staining to strong brown staining. e, f) Positive staining indicated by black arrows (scale bars=100 µm). A score was recorded for the staining confined to the macrophages (Macs), the epithelium (Epi) and for the overall slide. 10 different fields were scored on each slide and the average score was calculated for each slide. An average score of the scores for each of the two observers was then calculated. Example d–f) p-STAT1 and k–m) p-STAT3 staining of d, k) nonsmoking, e, l) smoking and f, m) COPD tissues are shown. Mean±sem g) p-STAT1 and n) p-STAT3 data are shown. Statistical significance was assessed using a Kruskal–Wallis test and Dunn's multiple comparison post-test. *: p<0.05 versus donor.
IHC staining for tyrosine phospho-STAT1 displayed a similar trend towards a small increase in smokers and COPD patients compared with nonsmokers but this did not reach significance. Staining was stronger in macrophages than epithelial cells across all three groups. A significant increase in tyrosine-phosphorylated STAT2 staining was observed in the macrophages of smokers compared with nonsmokers by IHC (score increased significantly from 1.93±0.16 to 2.57±0.19; p<0.05). This increase did not reach significance in epithelial cells or across the overall section and no such increase was seen in COPD patients. In contrast to the Western blotting data, no increase in tyrosine-phosphorylated STAT3 staining was seen in COPD patients compared with nonsmokers by IHC. There appeared to be a small increase in phospho-STAT3 in macrophages specifically from smokers compared with nonsmokers but this did not reach significance. No difference in phospho-STAT4 or phospho-STAT5 staining could be detected by IHC across the groups in either cell type (epithelial cells or macrophages) evaluated or the overall section. In contrast, we could only detect very low tyrosine-phosphorylated STAT6 staining by IHC and there were no significant differences between the groups.
To our knowledge, this is the first study to show increased phosphorylation of tyrosine on STAT1 in lung parenchymal samples taken from COPD patients compared with smokers without COPD and nonsmokers. This is particularly interesting as a recent genome-wide association study of a Norwegian cohort suggested that a polymorphism in the STAT1 gene may confer susceptibility to developing COPD [4]. Equally, we were able to show increased phosphorylation of STAT3 by Western blotting in COPD patients compared with nonsmokers. This finding is consistent with elevated levels of interleukin (IL)-6 that are seen in the induced sputum and lung tissue of COPD patients [5, 6] and the fact that IL-6 is recognised to activate STAT3 [7]. In addition, other cytokines, such as interferon-γ, IL-2, IL-12, IL-17 and IL-22, may either stimulate the JAK–STAT signalling pathway or be expressed as a consequence of activation of this pathway. The production of these cytokines is likely to be involved in the chronic cellular inflammation observed in COPD and the subsequent altered lung structure/function. Interestingly, we could not detect any phosphorylation of Ser727 on either STAT1 or STAT3. As these sites are not thought to be phosphorylated by JAK, it may imply that the activation of STATs we observe is specific to JAK signalling. These data may suggest that while the JAK–STAT pathway is involved in the pathogenesis of COPD, only certain STATs are involved. Therefore, the possibility exists to target specific STAT proteins rather than broad-spectrum inhibition, which may increase the side-effect liability. However, the influence of cigarette smoking on JAK–STAT activation, independent of disease status, remains a confounding factor.
While the results obtained by IHC did not entirely match the results obtained by Western blotting, the differences in results may simply be explained by the different protocols employed. For example, fixing the lung tissue in formalin before IHC can mask epitopes of the target proteins by crosslinking proteins and/or altering their tertiary structure [8], although steps were taken to minimise this and unmask the epitopes. Alternatively, the discrepancy in the results may be due to differences in antibody affinity when used for each technique. Apart from the increase in STAT2 activation in macrophages, no significant differences in STAT activation between the groups were detected by IHC. While we were able to show an increase in STAT3 phosphorylation by Western blotting in COPD patients compared with nonsmokers, our IHC results did not illustrate this. Intriguingly, Ruwanpura et al. [6] were only able to demonstrate an association between activation of STAT3 and inflammation, and not COPD status, when comparing COPD patients to “healthy” smokers using IHC. While this appears to support our data showing no difference in phosphorylated STAT3 staining between COPD patients and smokers, it suggests that as COPD patients display airway inflammation but nonsmokers should not, we should have observed a difference between these two groups. This suggests that the antibody in this case was more suitable for Western blotting than IHC or that the dynamic range for the two assays is different.
Increased phospho-STAT4 staining by IHC in smokers and COPD patients compared with nonsmokers has been reported previously by Di Stefano et al. [9]. While we used the same antibody, we were unable to replicate their finding. This could be due to differences in the general protocol or the samples employed. The previous paper used samples from patients with mild or moderate COPD [9], whereas the samples in our study were from a transplant programme and, therefore, the patients had end-stage, severe disease. It may be that STAT4 is only activated during the early stages of COPD rather than the later stages.
In summary, although there are limitations associated with the samples used for this study, the results suggest a role for the JAK–STAT pathway in severe COPD, and STAT1 and STAT3, in particular, warrant further investigation as targets for novel COPD therapeutics.
Footnotes
Conflict of interest: Disclosures can be found alongside the online version of this article at erj.ersjournals.com
- Received April 7, 2014.
- Accepted March 17, 2015.
- Copyright ©ERS 2015










