





Abstract
Intermittent hypoxia, the main stimulus of obstructive sleep apnoea (OSA), induces inflammation, leading to early atherosclerosis. Whether the cyclooxygenase (COX) pathway contributes to intermittent hypoxia-induced atherosclerosis remains to be determined.
We studied the effects of 8-weeks of intermittent hypoxia exposure on COX-pathway gene expression and atherosclerosis, and the influence of COX-1 inhibition by SC-560 on atherosclerosis progression in aortas of apolipoprotein E-/- mice. Urinary 11-dehydrothromboxane B2 (11-dTXB2) was assessed in 50 OSA subjects free of cardiovascular risk factor matched for age and body mass index with 25 controls, and 56 OSA with cardiovascular risk factor.
Intermittent hypoxia significantly increased atherosclerotic lesion sizes, mRNA levels of COX-1 and thromboxane synthase (TXBS). Lesion sizes correlated to COX-1 (r = 0.654, p = 0.0003) and TXBS (r = 0.693, p<0.0001) mRNA levels. COX-1 inhibition reduced lesion progression in intermittent hypoxia mice only (p = 0.04). Urinary 11-dTXB2 was similar in OSA subjects free of cardiovascular risk factor and controls, but was increased by 13% (p = 0.007) in OSA subjects with cardiovascular risk factor compared with those without.
Although OSA itself was not associated with increased urinary 11-dTXB2 concentration, the COX-1 pathway was activated in intermittent hypoxia-exposed mice and in OSA subjects presenting with cardiovascular risk factor, and may contribute to intermittent hypoxia-induced atherogenesis. COX-1 inhibition could be of clinical interest in the prevention of cardiovascular morbidity in OSA.
Introduction
Obstructive sleep apnoea (OSA) is characterised by recurrent episodes of partial or complete upper airway obstruction occurring during sleep, leading to chronic intermittent hypoxia (CIH). This hallmark is the main factor involved in cardiovascular remodelling in OSA [1]. In OSA patients, early signs of atherosclerosis correlated to hypoxia severity [2], even after adjustment for confounding factors. OSA is associated with increased cardiovascular morbidity and mortality, and is identified as an independent cardiovascular risk factor (CVRF) [3]. Moreover, exposure of apolipoprotein E-deficient (ApoE-/-) mice [4, 5] and C57BL/6J mice [6] to CIH accelerated atherogenesis progression.
Atherosclerosis is a chronic inflammatory disease. Among the many inflammatory mediators involved in atherogenesis, we previously demonstrated the contribution of arachidonic acid-derived metabolites in OSA patients. Notably, OSA patients exhibited elevated levels of leukotriene B4 [7, 8] and cysteinyl-leukotrienes [9] that were associated with vascular remodelling. However, the cyclooxygenase (COX)-dependant pathway of arachidonic acid metabolism was poorly studied in OSA patients, and its implication in CIH-induced atherogenesis is unknown.
Thromboxane A2 (TXA2) and prostacyclin (PGI2) are two COX-derived metabolites of arachidonic acid metabolism. TXA2 is predominantly generated by platelets through COX type 1 isoform (COX-1) and thromboxane synthase (TXBS). TXA2 is quickly metabolised to thromboxane B2 (TXB2) and 11-dehydrothromboxane B2 (11-dTXB2), two metabolites respectively quantifiable in plasma and urine. TXA2 binding on its TP receptors induces platelet activation, vasoconstriction, vascular smooth muscle cell proliferation and increases expression of adhesion molecules [10]. The production of PGI2 mainly depends on endothelial COX-2 and prostacyclin synthase (PGIS). PGI2 is rapidly metabolised to 6-keto-prostaglandin F1α (6-keto-PGF1α in plasma and 2,3-dinor-6-ketoprostaglandin F1α (PGI-M) excreted in urine. In contrast to TXA2, PGI2 binding on its IP receptors inhibits platelet aggregation and vasoconstriction, and reduces chemotaxis and expression of adhesion molecules [10]. Thus, TXA2 and PGI2 have antagonist properties.
A recent growing body of evidence suggests a major role of the COX pathway in the pathogenesis and progression of atherosclerosis. In fact, pharmacological inhibition of COX pathway [11, 12] or genetic deletion [13, 14] delayed atherosclerosis in different mouse models of atherosclerosis. Furthermore, the urinary 11-dTXB2/PGI2-M ratio is enhanced in elderly subjects with past history of serious cardiovascular event (stroke and myocardial infarction) [15]. As selective COX-2 inhibitors may have nonfavourable effects on this ratio and increase cardiovascular risk [16], the role of COX-1 inhibition has received less attention. In addition, data in OSA are very limited and conflicting, showing either an increased [17] or a decreased [18] urinary ratio 11-dTXB2/PGI2-M. Thus, the aim of the present study was to characterise the COX pathway in ApoE-/- mice exposed to CIH and in OSA patients, and to investigate its link with CIH-induced atherogenesis and OSA-associated early vascular remodelling.
Materials and methods
Experimental animal study
Male ApoE-/- mice (14 weeks old) were purchased from the Charles River Laboratories (L'Arbresle, France). All animal procedures were conducted in accordance with the European Convention for the Protection of Vertebrate Animals used for Experimental and Other Scientific Purposes (Council of Europe, European Treaties ETS 123, Strasbourg, March 18, 1986) and to the Guide for the Care and Use of Laboratory Animals (NIH Publication no. 85–23, revised 1996).
In a first series of experiments, mice were randomised to 8 weeks of CIH (cyclic 21–25% inspiratory oxygen fraction (FIO2), 60-s cycle for 8 h·day−1) or normoxic air as previously described (n = 15 in each group) [1]. In a second series of experiments, mice were exposed to 8 weeks of CIH or normoxic air (n = 20 in each group), and randomised to receive either placebo or the selective COX-1 inhibitor SC-560 (Interchim, Montluçon, France) (15 mg·kg−1 daily [12]) in their food, for the last 4 weeks of exposure. All mice were fed with a normal diet, ad libitum, during all experiments.
At the end of exposure, blood was collected under anaesthesia (ketamine/xylazine 100 mg·kg−1/10 mg·kg−1 by intraperitoneal injection) for haematocrit and lipid measurements. Entire aortas were harvested. Abdominal aortas were placed in RNAlater (Life Technologies, Villebon-sur-Yvette, France), frozen in liquid nitrogen and stored at -80°C until analysis. Immediately after their sampling, thoracic aortas were placed in Tyrode solution (137 mM NaCl, 2.7 mM KCl, 0.41 mM NaHPO4, 2 mM CaCl2, 5 μM MgCl2, 11.9 mM NaHCO3 and 5.5 mM glucose) for prostanoid secretion measurements.
Atherosclerotic lesion size quantification
Atherosclerotic lesions of aortic roots were studied by Oil-Red-O (Sigma Aldrich, Saint Quentin-Fallavier, France) staining, as previously described [5]. For each aorta, lipid deposition was quantified from five sections (8-μm thickness), separated by 80 μm from each other, using computer image analysis (NisElement; Nikon Instruments Inc., Melville, NY, USA).
Aortic secretion of prostanoids
Thoracic aortas were incubated for 15 min in Tyrode solution maintained at 37°C, aerated with 95% O2 and 5% CO2, in presence of calcium ionophore A23187 (Sigma Aldrich, Saint Quentin Fallavier, France) at 10−6 M. Supernatants were immediately frozen at -80°C for prostanoid measurement, while aortas were dried for measurement of dry tissue weight.
COX-pathway gene expression
Total mRNA was isolated from aorta using the RNeasy kit (Qiagen, Hilden, Germany) as previously described [19] and reverse-transcribed using Superscript II (Invitrogen, Carlsbad, CA, USA) with random hexamers according to the manufacturer's instructions. Quantitative TaqMan PCR was performed on a 7900 HT using primer/probe pairs designed with Assay-On-Demand (both Applied Biosystems; Life Technologies SAS, Saint Aubin, France) (online supplementary table S1). Data were normalised to 18S ribosomal protein mRNA and expressed as 2-ΔCT.
Clinical study
The local ethics committee approved this study according to the Declaration of Helsinki. All participants gave written informed consent.
113 newly diagnosed OSA patients were consecutively entered into the study between January 2007 and April 2011, as well as 25 controls. Patients were referred to the sleep laboratory of Grenoble University Hospital for symptoms suggesting OSA. Exclusion criteria were past history of stroke or myocardial infarction, known hypertension, and treatment with nonsteroidal anti-inflammatories, aspirin, steroids, antidiabetic, antihypertensive and lipid-lowering drugs.
Design
The specific contribution of CIH on 11-dTXB2 urinary concentrations was assessed in 25 controls carefully matched for age and body mass index (BMI) with 50 nonobese OSA patients (i.e. one healthy subject for two OSA subjects). All subjects were free of any known CVRFs. A sample size of 50 OSA patients and 25 controls was calculated as adequate to show an increase of 30% in urinary 11-dTXB2 excretion with at least 90% power and at the 5% significance level. This sample size calculation was based on preliminary data obtained on healthy subjects showing a mean concentration of 11-dTXB2 of 647 pg·mL−1 with a standard deviation of 287 pg·mL−1. As the dispersion of 11-dTXB2 was higher in patients with cardiovascular disease than in controls, we chose a mixed model including one healthy for every two OSA subjects.
The influence of CVRFs (which comprise obesity (BMI >30 kg·m−2); hypertension (clinical diastolic blood pressure (BP) >90 mmHg and systolic BP >140 mmHg); dyslipidemia (low-density lipoprotein (LDL) cholesterol >4.13 mmol·l−1 or association of total cholesterol >5.16 mmol·l−1 and high-density lipoprotein (HDL) cholesterol <1.03 mmol·l−1); smoking; and metabolic syndrome (defined by criteria of International Diabetes Federation [20])) on urinary excretion of 11-dTXB2 was evaluated in the whole cohort of 113 OSA subjects. Patients were stratified on the presence or not of one of these CVRFs.
The influence of continuous positive airway pressure (CPAP) on urinary 11-dTXB2 concentration was studied in 14 OSA subjects free of any CVRF and 21 OSA patients with CVRF(s) who were adherent to CPAP. CPAP adherence was defined as CPAP daily use for >4 h [9].
All subjects underwent an overnight polysomnography as described [9]. Sleep apnoea was defined as an apnoea/hypopnoea index (AHI) ≥5 events·h−1 of sleep and symptoms or respiratory disturbance index (RDI), including flow limitation episodes >15 events·h−1 [21].
Urine sample for 11-dTXB2 quantification and venous blood for biochemical measurements were collected at 07:00 h at the end of the nocturnal polysomnographic recordings and were stored at -80°C until later analysis.
Carotid ultrasonography was performed in 97 OSA patients and 24 controls as previously described [2].
Biological measurements
Plasma cholesterol, triglycerides, glucose and high-sensitivity C reactive protein (hsCRP) concentrations were determined using enzymatic colorimetric methods on a Dimension Vista analyser (Siemens AG, Erlangen, Germany). LDL cholesterol was calculated using the Friedewald formula (total cholesterol-HDL cholesterol-(triglycerides/5)). Urinary 11-dTXB2 of patients, TXB2 and 6-keto-PGF-1α in supernatants of mice were measured by liquid chromatography–tandem mass spectrometry (LC-MS/MS) as previously described [9, 22]. The limits of quantitation of TXB2 and 6-keto-PGF1α were 14 pg·mL−1 and 7.25 pg·mL−1, respectively, and their inter- and intra-assay coefficients of variation were <12% for both analytes.
Statistical analysis
Statistical analyses were performed using NCSS97 (Kaysville, Utah) or SAS (SAS 9.1, Cary, NC, USA) for the mixed model. Data were expressed as median and 10th and 90th percentiles. Normal distribution was tested using Kolmogorov–Smirnov nonparametric test. Comparisons between normoxic air and CIH mice were made using the t-test. Comparisons between OSA patients and controls matched for age and BMI (one control for two OSA subjects) were made using a mixed model. Noncontinuous variables were compared using Fisher’s test. Comparisons between more than three groups were made using the Kruskal–Wallis test and subsequent pairwise comparisons were made with the Bonferroni multiple-comparison test. For patients treated by CPAP, differences between baseline and post-CPAP values were analysed using a paired t-test or the Wilcoxon signed-rank test. Relationships between two variables were studied with single regression. A multiple linear regression analysis was performed, taking into account the variables that correlated with dependant variable urinary 11-dTXB2 in humans. A p-value of <0.05 was considered significant.
Results
Experimental animal study
Weight, lipid levels and haematocrit
After 8-weeks of exposure, bodyweight and plasma cholesterol levels were similar in CIH and normoxic air mice (table 1). Haematocrit was significantly higher in CIH mice compared with normoxic air mice (table 1).
Atherosclerotic lesion sizes
Atherosclerotic lesions on aortic roots were higher (p = 0.008) in CIH mice compared with normoxic mice (CIH 66 532 (32 741–163 224) versus normoxic air 37 675 (6798–75 596) μm2; p = 0.03). Lesion sizes correlated with plasma total cholesterol in normoxic air mice (r = 0.501, p = 0.04) but not in CIH mice.
COX-pathway gene expression
COX-1 and TXBS aortic mRNA levels were significantly increased in CIH group compared with normoxic air, whereas COX-2, PGIS, TP and IP receptors aortic mRNA levels were not significantly different between both groups (fig. 1a). mRNA levels of COX-1 and TXBS significantly correlated with lesion sizes (fig. 1b).

a) mRNA levels of COX-pathway genes in mice exposed to chronic intermittent hypoxia in the abdominal aorta. Regressions are shown between atherosclerotic lesion size and aortic mRNA levels of b) COX-1 and c) thromboxane synthase (TXBS). Data are expressed as fold-changes compared with normoxic mice (control). *: p<0.05 versus normoxic group. COX-1: cyclooxygenase type 1 isoform; COX-2: COX type 2 isoform; TXBS: thromboxane synthase; PGIS: prostacyclin synthase.
Aortic prostanoid secretion
A23187-stimulated 6-keto-PGF1α and TXB2 secretions and 6-keto-PGF1α/TXB2 ratio were similar in aorta from CIH and normoxic air mice (table 2). 6-keto-PGF1α and TXB2 concentrations were highly correlated (r = 0.842, p<0.0001).
Effects of COX-1 inhibition
In both normoxic air and CIH groups, treatment with SC-560 significantly decreased TXB2 and 6-keto-PGF1α aortic secretion versus placebo (table 2). There was no difference between groups for total cholesterol and bodyweight (table 2). Treatment with SC-560 significantly reduced lesion size by 35% in CIH mice whereas it had no effect in normoxic air mice (fig. 2).
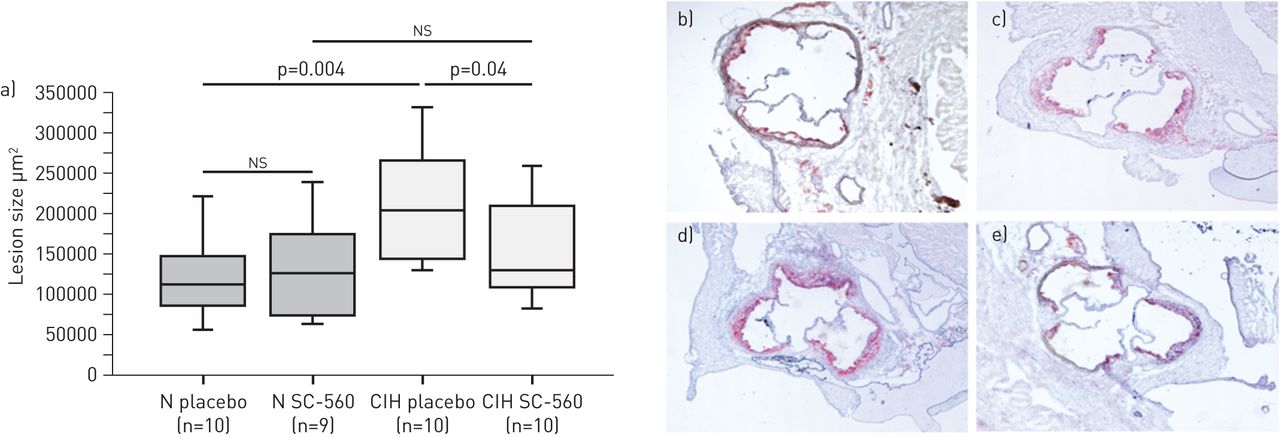
Effects of cyclooxygenase type 1 isoform (COX-1) inhibition on atherosclerosis in mice exposed to chronic intermittent hypoxia (CIH) or normoxia (N). a) Lesion sizes. Data are presented as interquartile range (boxes), data range (whiskers) and median (horizontal line). ns: nonsignificant. Representative photographs of Oil-Red-O staining for b) normoxia placebo, c) normoxia SC-560, d) CIH placebo and e) CIH SC-560.
Clinical study
11-dTXB2 and OSA
Baseline characteristics of the 25 controls and the 50 OSA patients matched for age and BMI are described in table 3. There was no significant difference for plasma insulin, hsCRP, HDL cholesterol, homeostatic model assessment insulin resistance index (HOMA-IR), BP, carotid intima-media thickness (IMT) and sex ratio. As expected, polysomnographic parameters were different between OSA and control groups. Plasma glucose, total and LDL cholesterol levels were significantly increased in OSA patients versus controls. Urinary 11-dTXB2 was not significantly different between OSA patients and controls (table 3).
11-dTXB2, OSA and cardiovascular risk factor
Clinical, biological and polysomnographic parameters of OSA patients with or without CVRFs are shown in table 4. The OSA group with CVRFs had polysomnographic parameters, BP, BMI, plasma triglycerides, total and LDL cholesterol, glucose, insulin and HOMA-IR higher than the OSA group free of CVRFs (table 4). Conversely, these two groups were similar with regard to age, sex ratio, carotid IMT, HDL cholesterol and hsCRP (table 4). Urinary 11-dTXB2 was significantly increased in OSA patients with CVRFs compared with OSA patients free of CVRFs (fig. 3a). In OSA patients with or without CVRFs, 11-TXB2 levels were similar in patients with mild-to-moderate (AHI <30 events·h−1) and severe OSA (AHI ≥30 events·h−1) (data not shown).

{kind=link}
{kind=link}
{kind=link}
a) Urinary 11-dehydrothromboxane B2 (11-dTXB2) concentrations in obstructive sleep apnoea (OSA) patients with (CVRF+) and without (CVRF-) known cardiovascular risk factor. b) Urinary 11-dTXB2 concentrations in OSA patients with or without carotid wall hypertrophy. Data are presented as interquartile range (boxes), data range (whiskers) and median (horizontal line).
To determine the CVRF involved in the increase of urinary 11-dTXB2, simple regressions were performed. Hypertension (r = 0.190, p = 0.05) and obesity (r = 0.242, p = 0.01) were weakly associated with increased urinary excretion of 11-dTXB2. In multiple regression model, obesity remained the sole independent predictive factor of urinary 11-dTXB2 (r = 0.257, p = 0.04).
Effect of CPAP treatment on urinary 11-dTXB2 concentration
CPAP treatment for at least 8 weeks significantly decreased AHI, RDI and respiratory arousal index, increased minimal nocturnal arterial oxygen saturation (SaO2) and mean nocturnal Sa,O2, and decreased the percentage of time spent with a mean SaO2 <90%. CPAP treatment induced no change on 11-dTXB2 urinary concentration in OSA without or with CVRF (online supplementary tables S2 and S3).
11-dTXB2, OSA and vascular remodelling
In OSA patients free of CVRF, vascular hypertrophy (defined by carotid IMT >0.8 mm) was associated with increased urinary 11-dTXB2 concentrations compared with subjects without vascular hypertrophy (p = 0.02) (fig. 3b).
Discussion
Our study demonstrated for the first time an activation of the COX pathway in ApoE-/- mice exposed to CIH and also in OSA patients with other CVRFs; this activation is associated with increased atherosclerotic lesions in mice and with early markers of atherosclerosis in OSA patients.
We showed that CIH mice had higher atherosclerotic lesions than normoxic air mice, suggesting that, in our model, CIH might have accelerated atherosclerosis development. This result is in accordance with previous works in ApoE-/- mice exposed to CIH from 2 to 4 weeks [4, 5] or in C57BL/6J mice exposed to CIH for 12 weeks [6].
In the present study, plasma cholesterol levels were not significantly different between normoxic air and CIH groups and atherosclerotic lesion sizes correlated with plasma cholesterol in normoxic air mice, but not in CIH mice. These data suggested that, in our model, CIH-induced atherosclerosis might be independent of lipid disorders, and contrast with previous works showing that concomitant exposure to a high-fat high-cholesterol diet and CIH aggravate both atherosclerosis and dyslipidemia in ApoE-/- mice [4] and in C57BL/6J mice [6, 23]. However, we recently showed that CIH also exerts pro-atherogenic effects through other contributing factors, notably the inflammatory process [5]. In agreement with this later hypothesis, our data showed an activation of the thromboxane pathway in CIH mice, as mRNA levels of COX-1 and TXBS were increased in aortic tissue of CIH mice. Furthermore, the correlation between these mRNA levels and atherosclerotic lesion size was consistent with direct effects of TXA2 on macrophages [24]. Unexpectedly, the aortic secretion of TXB2 and 6-keto-PGF1α, as well as the TXB2/6-keto-PGF1α ratio were similar in CIH and normoxic air mice. These results could be explained by the fact that we measured prostanoid secretion after aorta stimulation with A23187, and not the basal aortic production. A previous study performed in LDL r-knockout (r-KO) mice demonstrated that the acceleration of atherogenesis in response to high fat diet was associated with increased basal levels of TXB2 and 6-keto-PGF1α in the aortic arch [25], but measurements of prostanoids were performed by ELISA, which is not a specific method.
We chose to measure TXB2 and 6-keto-PGF1α upon aorta stimulation with A23187 to obtain detectable prostanoid levels by our analytical technique (LC-MS/MS), which is highly specific but probably less sensitive. However, in our model, A23187 appears to stimulate TXB2 and 6-keto-PGF1α release to the same extent, as both levels were highly correlated, which could explain the similar TXB2/6-keto-PGF1α ratio in aorta from CIH and normoxic air mice. We acknowledge that comparisons of basal TXB2 and 6-keto-PGF1α production and Western blot analysis of COX-1 and TXBS would have been of interest with regard to the increased mRNA levels of COX-1 and TXBS induced by CIH, and that their absence may represent a limitation of our study.
Moreover, treatment with the selective COX-1 inhibitor SC-560 during the last 4 weeks of CIH exposure reduced atherosclerosis progression in CIH mice, providing further evidence for a CIH-dependent activation of the COX pathway. We already demonstrated that 4 weeks of CIH exposure were sufficient to induce atherosclerosis in ApoE-/- mice [5], thus it was of interest to explore the effects of COX-1 inhibition on established atherosclerotic lesions. As previously described [12], treatment with SC-560 had no effect on established atherosclerotic lesions in normoxic air mice, although it was effective in inhibiting COX-1 pathway as assessed by the measurement of aortic TXB2 and 6-keto-PGF1α secretion. Collectively, these data demonstrated that in ApoE-/- mice, the atherogenic process was accelerated by CIH exposure, at least in part through COX-1 pathway activation. CIH activates leukocytes [26] and OSA patients display leukocyte activation [7, 8, 27]. By regulating the interaction between leukocytes, smooth muscle cells and endothelial cells, TXA2 promotes and PGI2 prevents the initiation and progression of atherogenesis [14].
To extend the thromboxane-pathway activation observed in CIH mice to OSA patients, we measured the urinary excretion of 11-dTXB2, a validated biomarker of systemic TXA2 production [10]. Urinary 11-dTXB2 concentrations of OSA patients free of CVRF were not different to those of controls carefully matched for age and BMI, two major confounding factors often present in studies focused on the underlying inflammation associated to OSA. These data suggested that OSA itself was not associated with an increased urinary 11-dTXB2 excretion, an hypothesis confirmed by the observation that CPAP treatment had no influence on urinary 11-dTXB2 level. However, increased urinary 11-dTXB2 concentrations have previously been described in patients with cardiovascular diseases including obesity [28] and hypertension [29]. Consistent with these data, we showed that OSA patients with an associated CVRF had higher urinary concentrations of 11-dTXB2 than OSA patients free of a known CVRF. Furthermore, among the studied CVRFs, obesity was the sole independent predictor of urinary 11-dTXB2 excretion, which is in agreement with a previous study demonstrating an increased urinary 11-dTXB2 excretion in obese females [28]. This result provided further evidence for the major role of obesity in arachidonic acid metabolism activation in OSA patients, as we previously described for the 5-lipooxygenase pathway [9]. Finally, urinary 11-dTXB2 excretion was associated with carotid wall hypertrophy in OSA patients. Again, these findings were consistent with the proliferative effects of TXA2 on vascular smooth muscle cells [10], although it must be kept in mind that vascular remodelling is a complex process implying many mediators and that COX pathway activation might not be the sole mechanism of vascular remodelling in OSA patients with CVRFs.
In conclusion, our study showed an activation of the COX-1 pathway in ApoE-/- mice exposed to CIH, which contributed to the acceleration of the atherogenic process induced by this stimulus. Such an activation of the COX-1 pathway was also found in OSA patients with associated CVRFs. This study extends the activation of the arachidonic acid metabolism previously described for the leukotriene pathway [7, 9] to the COX-1 pathway in OSA in relation to vascular remodelling. These findings open the field to the interest of new pharmacological approaches (dual COX and 5-lipoxygenase inhibitor [30]) in the prevention of cardiovascular morbidity in OSA patients.
Acknowledgments
The authors thank Nathalie Arnold (CHU, Hôpital A. Michallon, Pôle rééducation et Physiologie, BP217, Grenoble, France) for statistical analyses, Jean-François Jourdil, Karine Scalabrino, Cécile Girard (CHU, Hôpital A. Michallon, Laboratoire de Pharmacologie, BP217, Grenoble) and Sandrine Cachot (INSERM, U1042, Grenoble) for expert technical assistance.
Footnotes
This article has supplementary material available from www.erj.ersjournals.com
Support statement: This study was supported by a grant from “le vivier de la recherche médicale” from Grenoble University of Medicine, PHRC 2010, ResMed Foundation, Mairie de Paris (“Research in Paris”), The French-Swedish Foundation and the Swedish Heart and Lung Foundation.
Conflict of interest: None declared.
- Received June 18, 2012.
- Accepted September 26, 2012.
- ©ERS 2013
References










