





Abstract
Intravenous administration of activated protein C (APC) inhibits coagulation and inflammation in the lungs of humans and animals. Investigations in rodents demonstrated that direct intrapulmonary delivery of APC also exerts anticoagulant and anti-inflammatory effects. The effect of intrabronchial administration of recombinant human (rh)APC on lipopolysaccharide (LPS)-induced haemostatic and inflammatory alterations in the bronchoalveolar space of humans was studied.
Eight subjects received rhAPC via intrabronchial instillation by bronchoscope, while in a contralateral subsegment subjects received saline; all subjects were challenged bilaterally with LPS in the same lung subsegments. Four additional subjects received rhAPC (75 μg), with saline as a control in the contralateral subsegment, while they were bilaterally “challenged” with saline. After 6 h a bronchoalveolar lavage was performed and coagulation and inflammatory parameters were measured.
rhAPC enhanced LPS-induced coagulation activation in the bronchoalveolar space, when compared with the control side. In addition, rhAPC amplified LPS-induced pro-inflammatory responses, as indicated by higher concentrations of cytokines and chemokines. rhAPC alone did not have procoagulant or pro-inflammatory effects.
Locally administered rhAPC has unexpected procoagulant and pro-inflammatory effects in LPS-challenged lung subsegments. These data argue against a role for intrapulmonary delivery of rhAPC as a treatment strategy for lung inflammatory disorders in humans.
Introduction
The airways represent a body site in which procoagulant and anticoagulant mechanisms can be initiated and regulated locally [1, 2]. Activated protein C (APC), an important anticoagulant, has been shown to have a reduced activity in the bronchoalveolar space in various lung inflammatory diseases [3–11]. Diminished APC levels may contribute to the disturbed haemostatic equilibrium in the lungs of patients suffering from these conditions, shifting the physiological anticoagulant environment into a net procoagulant state. Besides its anticoagulant effects, APC has been implicated in a variety of anti-inflammatory and/or cytoprotective effects, characterised by downregulation of pro-inflammatory pathways, upregulation of anti-inflammatory pathways and inhibition of neutrophil activity [12, 13].
We recently demonstrated that intrabronchial instillation of lipopolysaccharide (LPS), a constituent of the cell wall of Gram-negative bacteria, can reproduce haemostatic alterations found in inflammatory lung disorders, including a reduction in APC levels in the lungs of healthy volunteers [14, 15]. This human model of intrabronchial LPS delivery has also been used to study the in vivo effects of intravenous recombinant human (rh)APC treatment on pulmonary inflammation and coagulation [15, 16]. Intravenous rhAPC reduced LPS-induced neutrophil accumulation in the bronchoalveolar space [16] and inhibited local activation of coagulation [15]. Concurrently, LPS-induced suppression of fibrinolysis was partially prevented by intravenous rhAPC [15].
When administered intravenously, the strong anticoagulant effects of rhAPC may contribute to serious systemic side-effects such as intracranial haemorrhages [17–20], which may be circumvented by local administration of rhAPC. Although preclinical animal studies have shown that administration of APC via the airways exerts local anti-inflammatory and anticoagulant effects [21–24], no studies exist that have evaluated the effects of direct rhAPC administration into the lungs of humans. The present study, therefore, sought to determine the feasibility of local, intrabronchial administration of rhAPC to inhibit LPS-induced lung inflammation and coagulation in humans.
Methods
Study subjects and materials
12 nonsmoking males (age 23.2±1.1 years) were enrolled. Screening, consisting of a questionnaire, physical examination, routine blood and urine investigation, ECG and spirometry, did not reveal any abnormality. The protocol was approved by the institutional Medical Ethics Review Committee. All subjects provided written informed consent before enrolment. LPS was derived from Escherichia coli O:113 (Reference Endotoxin, CC-RE-Lot-3, National Institutes of Health, Bethesda, MD, USA). rhAPC (drotrecogin alfa (activated); Eli Lilly, Indianapolis, IN, USA) was purchased from a commercial supplier.
Study design
The study was set-up as a single-blind intervention study with a dose-escalation and a follow-up phase. A bilateral challenge of two contralateral lung subsegments was performed by intrabronchial instillation of LPS (4 ng·kg−1) by bronchoscopy, diluted in 10 mL of sterile saline. In one lung subsegment the instillation of LPS was followed immediately by administration of rhAPC (in 10 mL) in the same lung subsegment, whereas in the contralateral lung subsegment LPS administration was followed by instillation of 10 mL saline alone. The subjects were randomised to the left or right lung for rhAPC instillation. In the dose-escalating phase of the study, five-fold increases in the rhAPC dose were intended to be given in cohorts of four subjects per dose, starting at 15 μg. The predefined primary end-point was to reach a 30% decrease in thrombin-antithrombin complex (TATc) concentrations in the rhAPC-treated lung 6 h after intrabronchial LPS challenge relative to the contralateral LPS-challenged side; this end-point was based on the capacity of intravenous rhAPC to inhibit the rise in LPS-induced TATc concentrations in bronchoalveolar lavage fluid (BALF) by approximately 30% in our previous study [15]. Per study design, the sample size of the rhAPC dose group that would reach this primary end-point would be increased to a total of 12 subjects in the follow-up phase of the study. After completion of the first two cohorts in the dose-escalation phase (15 and 75 μg; n=4 for each dose), we decided to stop the study because it was considered highly unlikely that the primary study end-point could be reached. Instead, four additional subjects were included who received 75 μg rhAPC in one lung subsegment without previous intrabronchial LPS challenge; in these volunteers saline was given as a control in the contralateral lung subsegment.
Methods
A bilateral bronchoalveolar lavage (BAL) was performed 6 h post-challenge in a standardised fashion [14, 25]. Citrated and heparinised blood was taken by venipuncture before the first (t=0 h) and the second (t=6 h) bronchoscopy. The assays used are described in the online supplementary material.
Statistical analysis
Values are expressed as mean±sem. Paired t-tests were used to establish significance between datasets. p-values <0.05 were considered statistically significant.
Results
Clinical signs and systemic inflammatory response
Instillation of LPS in two contralateral lung subsegments (combined with rhAPC in one lung segment) was well tolerated. A modest rise in body temperature was recorded 6 h after instillation of LPS (from 36.0±0.2°C at t=0 h to 36.6±0.1°C at t=6 h; p<0.05). Intrabronchial LPS caused a rise in blood neutrophil count (from 2.3±0.2×109 to 6.6±0.8×109 cells·L−1; p<0.001), while in the saline-challenged group no significant rises in neutrophils counts were seen (from 2.0±0.2×109 to 4.1±0.8×109 cells·L−1). Finally, intrabronchial instillation of either LPS or saline, with addition of rhAPC in one lung subsegment, was not associated with changes in plasma concentrations of TATc, D-dimer, tumour necrosis factor (TNF)-α or interleukin (IL)-6 (data not shown).
Bronchoalveolar APC concentrations
6 h after intrabronchial instillation of rhAPC, APC was measurable in all BALF samples obtained from the APC-treated side: APC levels were 0.33±0.14 and 1.60±0.57 ng·mL−1 in LPS-challenged subjects treated with 15 and 75 μg rhAPC, respectively (p=0.07 and p<0.05 for lung subsegments treated with 15 and 75 μg rhAPC versus control lung subsegments; fig. 1a and b). In the saline-challenged group treated with 75 μg rhAPC, BALF APC levels were 3.2±1.0 ng·mL−1 (p<0.05; fig. 1c). APC levels in the 75 μg rhAPC-challenged subsegments were not different between LPS-challenged and saline-challenged subjects. APC could not be detected in any of the control lung subsegments (i.e. those not administered with rhAPC).

Activated protein C (APC) concentrations in bronchoalveolar lavage samples. Healthy subjects (n=4 per group) were challenged bilaterally in a lung subsegment via a bronchoscope with lipopolysaccharide (LPS; 4 ng·kg−1 body weight) (a and b) or saline (c). Directly thereafter subjects received recombinant human (rh)APC 15 μg (a) or 75 μg (b and c) dissolved in 10 mL of saline in one subsegment and 10 mL of saline in the other lung subsegment. Bilateral bronchoalveolar lavage was performed 6 h after challenge. APC levels were undetectable in any of the control lung subsegments not administered with rhAPC. *: p<0.05.
Intrabronchial rhAPC enhances bronchoalveolar coagulation activation after LPS challenge
The intrabronchial concentrations of TATc have been used to determine the extent of bronchoalveolar coagulation activation in patients with acute lung injury and/or pneumonia [7, 8, 10]. Intrabronchial instillation of LPS in healthy humans is also associated with a rise in TATc levels in BALF [14, 15]. An earlier study showed that intravenously administered rhAPC could reduce LPS-induced elevations in TATc concentrations in BALF by approximately 30% [15]. The main objective of the current study was to determine whether intrabronchially instilled rhAPC was able to inhibit LPS-induced TATc elevation in BALF and, if so, whether this anticoagulant effect was accompanied by anti-inflammatory effects. Considering the results of our previous investigation using intravenous rhAPC [15], we predefined the primary end-point of the current study as establishing the intrabronchial rhAPC dose that induced a 30% reduction in LPS-induced TATc concentrations in BALF. We chose to administer LPS in two contralateral lung subsegments, thereby eliminating inter-individual variation in LPS responsiveness and allowing every subject to serve as his own control with regard to rhAPC effects. Much to our surprise, intrabronchially instilled rhAPC, given at either 15 μg or 75 μg, induced an increase, rather than a reduction, in BALF TATc concentrations after LPS challenge when compared with the contralateral side not treated with rhAPC (rhAPC 15 μg: 13.5±5.2 versus 9.8±4.8 μg·L−1 at the contralateral side, p<0.01, fig. 2a; rhAPC 75 μg: 19.4±4.2 versus 2.9±0.8 μg·L−1 at the contralateral side, p<0.05, fig. 2b). As these results made it highly unlikely that we could reach our predefined end-point, we decided not to proceed with another five-fold increase in rhAPC dose but, instead, added an extra study group without LPS challenge, seeking to determine whether intrabronchial rhAPC per se elicited a procoagulant response. Thus, subjects received a bilateral challenge with saline, combined with rhAPC (75 μg) on one side. These studies revealed no procoagulant effect of rhAPC: BALF TATc levels were similar in lung subsegments instilled with rhAPC (1.6±1.4 μg·L−1) when compared with the contralateral side (2.4±1.2 μg·L−1; fig. 2c). Protein C activity, total and free protein S, soluble thrombomodulin, protein C inhibitor and von Willebrand factor antigen concentrations were all below detection limits in BALF.

Intrabronchial instillation of recombinant human activated protein C (rhAPC) augments lipopolysaccharide (LPS)-induced coagulation in the bronchoalveolar space. Healthy subjects (n=4 per group) were challenged bilaterally in a lung subsegment via a bronchoscope with LPS (4 ng·kg−1 body weight) (a and b) or saline (c). Directly thereafter subjects received rhAPC 15 μg (a) or 75 μg (b and c) dissolved in 10 mL of saline in one subsegment and 10 mL of saline in the other lung subsegment. Thrombin-antithrombin complex (TATc) levels were measured in bronchoalveolar lavage samples obtained 6 h after challenge. *: p<0.05; **: p<0.01.
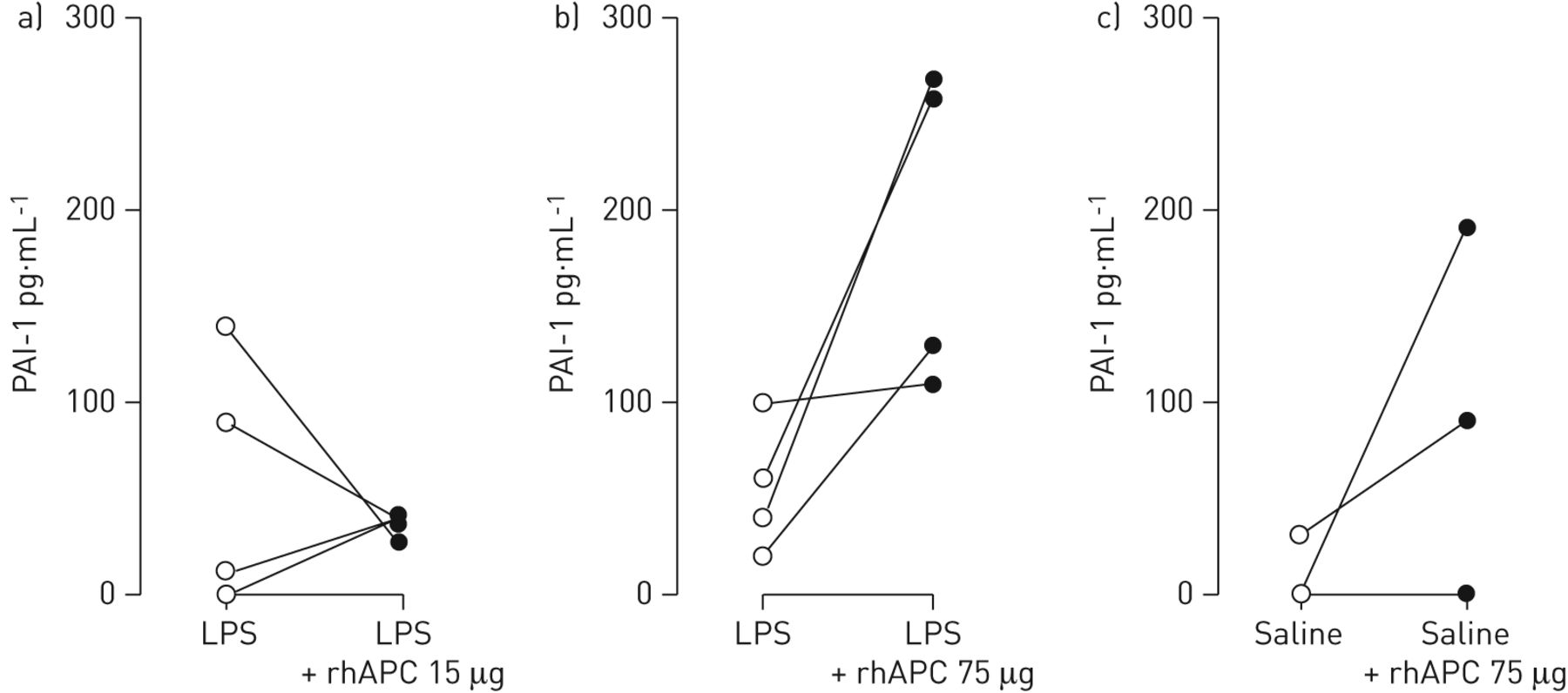
Effect of intrabronchial rhAPC on fibrinolysis after LPS challenge
Evidence derived from in vitro investigations indicates that APC may stimulate fibrinolysis by inhibiting plasminogen activator inhibitor type 1 (PAI-1) [26]. We measured levels of PAI-1, tissue-type plasminogen activator (tPA) and plasmin-α2-antiplasmin complexes. While intrabronchial rhAPC given at a dose of 15 μg did not influence LPS-induced PAI-1 release in BALF (p=0.57; fig. 3a), rhAPC administered at 75 μg increased LPS-induced PAI-1 BALF levels in three out of the four subjects (192.5±42.1 versus 55.0±17.1 pg·mL−1 at the contralateral side; p=0.07; fig. 3b). Notably, intrabronchial rhAPC also tended to increase PAI-1 levels in BALF from subjects who did not receive LPS (70.0±45.3 versus 7.5±7.5 pg·mL−1 at the contralateral side; p=0.26; fig. 3c). Neither LPS nor rhAPC influenced BALF concentrations of plasmin-α2-antiplasmin complexes, whereas tPA levels were below detection limit in all BALF samples (data not shown).

Impact of intrabronchial recombinant human activated protein C (rhAPC) on lipopolysaccharide (LPS)-induced release of plasminogen activator inhibitor type I in the bronchoalveolar space. Healthy subjects (n=4 per group) were challenged bilaterally in a lung subsegment via a bronchoscope with LPS (4 ng·kg−1 body weight) (a and b) or saline (c). Directly thereafter subjects received rhAPC 15 μg (a) or 75 μg (b and c) dissolved in 10 mL of saline in one subsegment and 10 mL of saline in the other lung subsegment. Plasminogen activator inhibitor type 1 (PAI-1) levels were measured in bronchoalveolar lavage samples obtained 6 h after challenge.
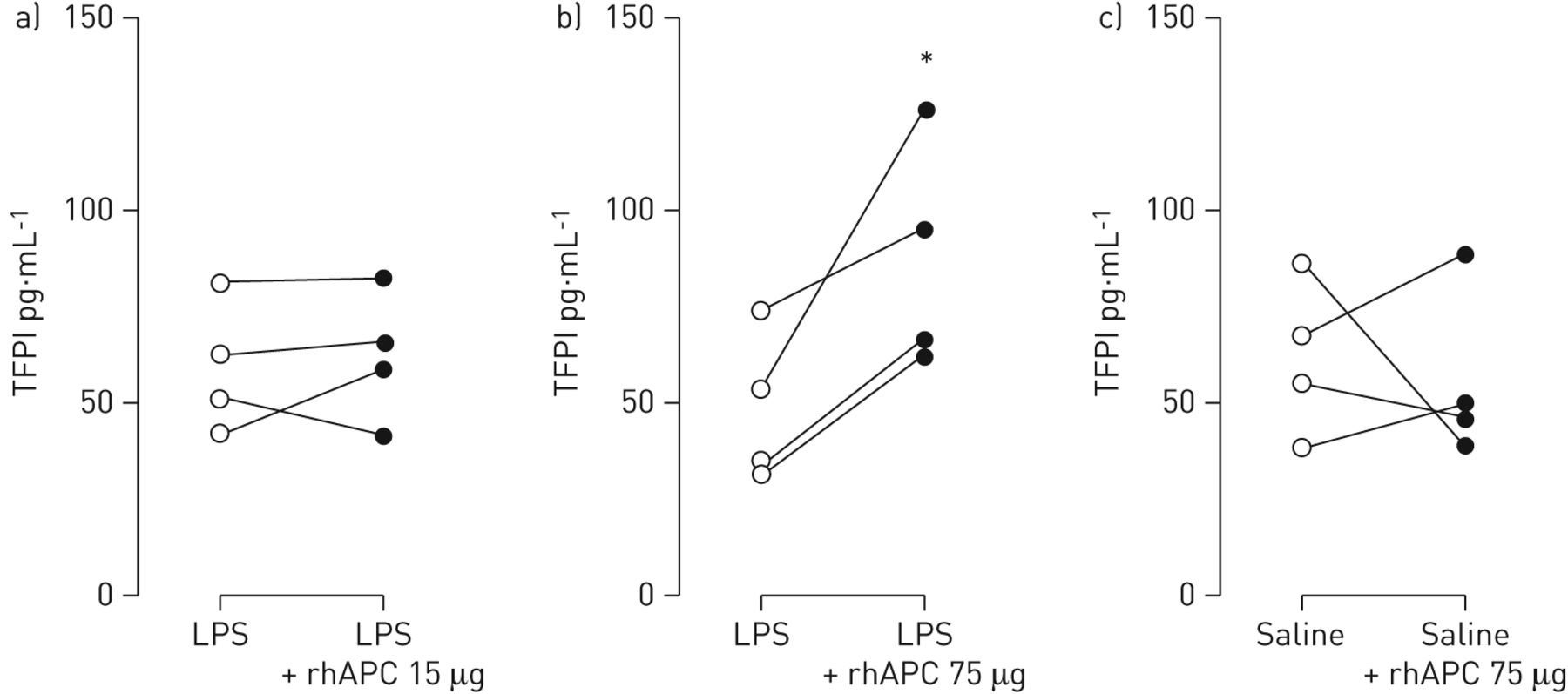
Intrabronchial rhAPC is associated with enhanced concentrations of tissue factor pathway inhibitor after LPS challenge
Recent data suggest that APC can have a procoagulant effect by virtue of its proteolytic activity. APC was reported to shed the Kunitz-1 domain from tissue factor pathway inhibitor (TFPI), resulting in increased tissue factor activity and, consequently, coagulation activation [27]. We measured levels of TFPI in BALF. While intrabronchial rhAPC given at a dose of 15 μg did not influence LPS-induced TFPI release in BALF (fig. 4a), rhAPC administered at 75 μg increased LPS-induced TFPI BALF levels in all four subjects (48.1±9.9 versus 87.9±14.8 pg·mL−1 at the contralateral side; p<0.05; fig. 4b). Intrabronchial rhAPC did not change levels of TFPI in BALF from subjects who did not receive LPS (fig. 4c).
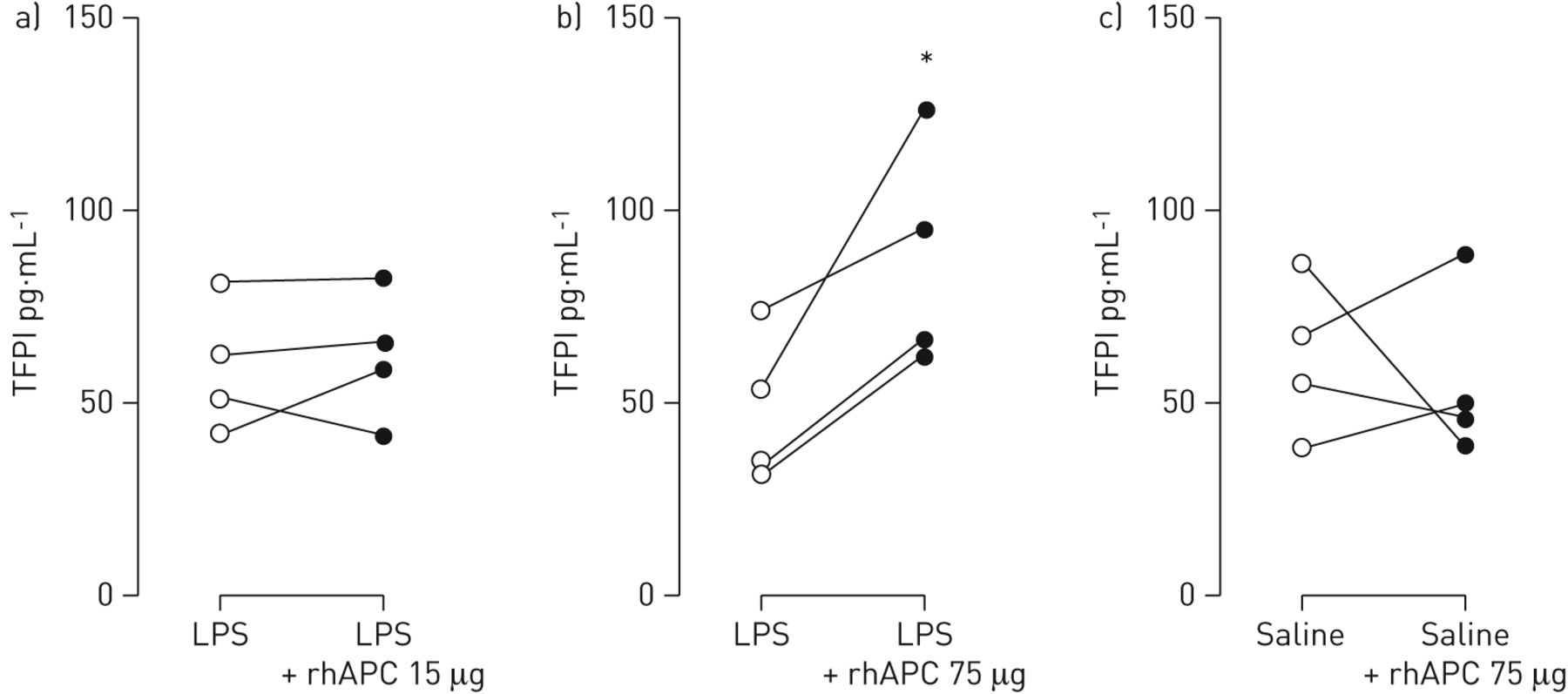
Impact of intrabronchial recombinant human activated protein C (rhAPC) on lipopolysaccharide (LPS)-induced release of tissue-factor pathway inhibitor in the bronchoalveolar space. Healthy subjects (n=4 per group) were challenged bilaterally in a lung subsegment via a bronchoscope with LPS (4 ng·kg−1 body weight) (a and b) or saline (c). Directly thereafter subjects received rhAPC 15 μg (a) or 75 μg (b and c) dissolved in 10 mL of saline in one subsegment and 10 mL of saline in the other lung subsegment. Tissue factor pathway inhibitor (TFPI) levels were measured in bronchoalveolar lavage samples obtained 6 h after challenge. *: p<0.05.
Intrabronchial rhAPC increases leukocyte recruitment after LPS administration
Intravenous rhAPC has been reported to reduce neutrophil recruitment into the bronchoalveolar space upon intrabronchial instillation of LPS in healthy humans [16]. In contrast, intrabronchial rhAPC treatment increased the total number of white blood cells in BALF 6 h after bilateral LPS challenge, although the differences with the contralateral control side did not reach statistical significance (APC 15 μg: 33.6±14.6×104 versus 18.6±4.1×104 cells·mL−1 at the contralateral side, p=0.25, fig. 5a; APC 75 μg: 52.0±22.4×104 versus 11.1±1.5×104 cells·mL−1 at the contralateral side, p=0.13, fig. 5b). The rhAPC-induced increase in total cell number in lung subsegments challenged with LPS was due to a rise in the number of neutrophils (APC 15 μg: 16.8±9.0×104 versus 7.5±2.7×104 neutrophils·mL−1, p=0.25, fig. 5d; APC 75 μg: 34.1±15.8×104 versus 1.5±0.6×104 neutrophils·mL−1, p=0.13, fig. 5e). Intrabronchial rhAPC did not affect cell counts in lung subsegments not challenged with LPS (fig. 5c and f).

Impact of intrabronchial recombinant human activated protein C (rhAPC) on the lipopolysaccharide (LPS)-induced increase in total leukocyte and neutrophil counts in the bronchoalveolar space. Healthy subjects (n=4 per group) were challenged bilaterally in a lung subsegment via a bronchoscope with LPS (4 ng·kg−1 body weight) (a and b) or saline (c). Directly thereafter subjects received rhAPC 15 μg (a) or 75 μg (b and c) dissolved in 10 mL of saline in one subsegment and 10 mL of saline in the other lung subsegment. White blood cell counts (WBCs) and neutrophil counts were determined in bronchoalveolar lavage samples obtained 6 h after challenge.
Intrabronchial rhAPC enhances the local release of several cytokines and chemokines after LPS instillation
To investigate the effect of local rhAPC administration on LPS-induced inflammatory mediator release we measured BALF concentrations of 48 cytokines, chemokines and growth factors. The results of these analyses are shown in online supplementary table S1. In the lower rhAPC dose cohort (15 μg), rhAPC significantly enhanced LPS-induced release of IL-6, CCL8 and CXCL6 while not influencing the levels of other mediators (online supplementary table S1). In the rhAPC 75 μg cohort, rhAPC significantly enhanced LPS-induced TNF-α, CCL4, CCL8 and TNF-related apoptosis-inducing ligand (TRAIL) concentrations (fig. 6 and online supplementary table S1). In addition, IL-1β, CCL14a, CCL20, CXCL7, CXCL9 and CXCL11 showed a strong trend toward higher levels in this higher rhAPC dose cohort (p=0.052, p=0.081, p=0.078, p=0.075, p=0.092 and p=0.074, respectively; fig. 6 and online supplementary table S1). rhAPC given at 75 μg without LPS did not induce the release of inflammatory mediators in the lung, with the exception of a modest rise in CCL15 concentrations (online supplementary table S1). Taken together, these results show that intrabronchial instillation of rhAPC in LPS-challenged lung subsegments enhanced the release of multiple pro-inflammatory mediators.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Intrabronchial instillation of recombinant human activated protein C (rhAPC) augments lipopolysaccharide (LPS)-induced release of inflammatory mediators in the bronchoalveolar space. Healthy subjects (n=4 per group) were challenged bilaterally in a lung subsegment via a bronchoscope with LPS (4 ng·kg−1 body weight). Directly thereafter subjects received rhAPC 75 μg dissolved in 10 mL of saline in one subsegment and 10 mL of saline in the other lung subsegment. Mediator levels were measured in bronchoalveolar lavage samples obtained 6 h after challenge. TNF: tumour necrosis factor; IL: interleukin; TRAIL: TNF-related apoptosis-inducing ligand. *: p<0.05; **: p<0.01.
Discussion
Several preclinical studies have shown that APC may protect the lung from injury caused by inflammation [28]. Moreover, intravenous administration of rhAPC was reported to exert anticoagulant and anti-inflammatory effects in the bronchoalveolar space of healthy humans challenged with LPS in a lung subsegment by bronchoscope [15, 16]. Animal studies demonstrated that intrabronchially instilled APC exerts local anticoagulant and anti-inflammatory effects [21–24, 29–32]. Considering the bleeding risk associated with intravenous APC, we here for the first time investigated the potential of intrabronchial rhAPC administration to inhibit coagulation and inflammation in the human lung, arguing that this route of delivery would circumvent serious side-effects [17–20]. For this we used the established human model of lung inflammation induced by intrabronchial instillation of LPS via a bronchoscope [14–16, 25]. The main finding of our study was remarkable: intrabronchially administered rhAPC induced a procoagulant and pro-inflammatory response in lung subsegments challenged with LPS rather than the expected inhibitory effects on coagulation and inflammation. Our results from subjects not challenged with LPS via the airways further suggest that rhAPC augments LPS effects in the human lung, while not eliciting detectable inflammation or coagulation per se.
Our study had a small sample size. As a consequence, although rhAPC clearly enhanced the LPS-induced release of biomarkers of coagulation and inflammation, the differences with the placebo control side often did not reach statistical significance. However, the current study was designed to demonstrate an inhibitory effect of rhAPC on bronchoalveolar coagulation and inflammation. After completion of the rhAPC 75 μg cohort, we considered it unlikely that the predefined end-point (i.e. a 30% reduction in BALF TATc levels) could be reached. Therefore, we decided to prematurely terminate the study. In addition, we considered it not ethical to enlarge the sample size of our cohorts in order to strengthen the statistical power of our study, on the basis that procoagulant and pro-inflammatory effects of locally administered rhAPC would not be of clinical value for patient groups and wishing to avoid exposing additional healthy subjects to invasive procedures.
In the earlier study, in which intravenous rhAPC was found to inhibit coagulation and neutrophil influx in the same human LPS-induced lung inflammation model [15, 16], APC concentrations in BALF were approximately 15 ng·mL−1 as measured 2 h after discontinuation of the rhAPC infusion (24 μg·kg−1·h−1) [15]. Although BALF APC levels detected after intravenous rhAPC infusion cannot be readily translated to a feasible rhAPC dose to be administered in the bronchoalveolar space, we conservatively started the dose-escalation study with 15 μg of intrabronchial rhAPC, taking into account the dilution caused by the BAL procedure and an estimated recovery of 50–60%. BALF APC levels measured in the current study, 6 h after instillation, were considerably lower than in the previous study using intravenous APC administration [15] (mean levels of 0.33 ng·mL−1 and 1.60 ng·mL−1 in the rhAPC 15 μg and 75 μg cohorts, respectively). Arguably, BALF APC concentrations were higher directly after rhAPC administration. Clearance of APC from the bronchoalveolar space is difficult to investigate in humans in light of the invasive procedure needed; nonetheless, evidence suggests that APC is slowly cleared from the alveolar compartment. Indeed, while the majority of patients with sepsis who are treated with intravenous rhAPC do not have detectable APC in plasma 2 h after discontinuation of the infusion [33], APC could still be recovered from the BALF of healthy humans in whom a similar rhAPC infusion was stopped 2 h earlier [15]. Hence, although BALF APC concentrations measured in the present (intrabronchial rhAPC) and previous (intravenous rhAPC) [15] investigation cannot be compared easily, these combined data suggest that the APC levels achieved in BALF here were likely within the same or lower range than those attained after intravenous rhAPC infusion.
How can the apparently differential effects of intravenous rhAPC [15] and intrabronchial rhAPC in LPS-induced human lung inflammation be explained? APC can exert anti-inflammatory, anti-apoptotic and barrier protective signals in endothelial cells via protease-activated receptor (PAR)1 by a mechanism that requires binding of APC to the endothelial cell protein C receptor (EPCR) [34]. Cytoprotective anti-inflammatory APC-PAR1 signalling has also been demonstrated in other cell types, in particular dendritic cells (EPCR dependent [35]) and macrophages (CD11/CD18 dependent [35, 36]). Furthermore, APC directly binds to activated α3β1, α5β1 and αVβ3 integrins and this interaction was essential for APC-induced inhibition of neutrophil extravasation into the bronchoalveolar space of mice [37]. These “cytoprotective” and/or anti-inflammatory APC effects in vivo likely depend on which cell types are affected and thereby on the route of rhAPC administration. Furthermore, the distribution of receptors involved in these APC effects may vary in different body compartments. In addition, recent data suggest that APC can have a procoagulant effect by virtue of its proteolytic activity; APC was reported to shed the Kunitz-1 domain from TFPI, resulting in increased tissue factor activity and, consequently, coagulation activation [27]. In this study we measured TFPI levels in BALF and showed that administration of rhAPC in the 75 μg cohort was associated with significantly enhanced TFPI levels. We hypothesise that increased levels of TFPI may have been a result of the pro-inflammatory state due to rhAPC administration. It should be noted that the TFPI assay used detects both full-length and cleaved TFPI. Hence, it is possible that the elevated TFPI levels were the result of cleavage of the intact protein by rhAPC, thereby contributing to a procoagulant effect. We do not have a clear explanation for the discrepancy between our current findings and earlier reports on the effect of intrapulmonary delivery of APC in mice [21–24], although differences in the exact method of administration, the dose and species studied may be involved.
Our study does not provide a clear mechanism underlying the unexpected pro-inflammatory and procoagulant effect of rhAPC in the human lung. We tried to reproduce the effects of APC in a mixed cell culture system using respiratory epithelial cells (A549) and either primary human alveolar macrophages or macrophage-like THP-1 cells but were unsuccessful (data not shown).
In conclusion, this is the first study on the effects of intrabronchially administered rhAPC in a controlled lung inflammation model in humans. Our data show that locally administered rhAPC has procoagulant and pro-inflammatory effects in lung segments exposed to LPS. These data argue against a role for intrapulmonary delivery of rhAPC as a treatment strategy for lung inflammatory disorders in humans.
Acknowledgments
The authors thank A.F. de Vos, D. Kruijswijk and D.C. Blok (all Center for Experimental and Molecular Medicine, Academic Medical Center/University of Amsterdam, Amsterdam, The Netherlands) for their assistance during the study days, G. van Mierlo (Dept of Immunopathology, Sanquin Research at CLB, Amsterdam, The Netherlands), W.F. Kopatz, M.A. Weijne, L.M. Leverink and J.A. Marquart (all Dept of Experimental Vascular Medicine, Academic Medical Center/University of Amsterdam, The Netherlands) for assistance in performing the coagulation measurements and E.M. Kemper, in-hospital pharmacist (Dept of Pharmacology, Academic Medical Center/University of Amsterdam, The Netherlands) for assistance in preparation of the study medication.
Footnotes
This article has supplementary material available from www.erj.ersjournals.com
Clinical trial: This study is registered at www.clinicaltrials.gov with identifier number NCT00943267 and at www.trialregister.nl with identifier number NTR1544.
Support statement: This study was supported by research grants from ZonMW (to L.M. Kager, grant number: 92003504), the Netherlands Asthma Foundation (J.D. de Boer; project 3.2.08.009) and the Stichting BeGeTu (to L.M. Kager).
Conflict of interest: None declared.
- Received April 5, 2012.
- Accepted September 29, 2012.
- ©ERS 2013
References










