





Abstract
The pathobiology of chronic obstructive pulmonary disease (COPD) is not completely understood. The aim of this study was to assess the expression of hypoxia inducible factor (HIF)-1α in lung tissue from patients with COPD/emphysema.
Lung tissue samples from 26 patients were included in this study. Seven samples were obtained from patients with normal lung function, the remainder of the samples were taken from patients with moderate COPD (n = 6; stage I and II Global Initiative for Chronic Obstructive Lung Disease classification) and severe COPD (n = 13; stage III and IV).
We analysed mRNA and protein expression in the lung tissue samples and found that: 1) HIF-1α and histone deacetylase 2 proteins were significantly decreased and were correlated; 2) HIF-1α and vascular endothelial growth factor (VEGF) proteins, and forced expiratory volume in 1 s % predicted were correlated in all patients; 3) the changes in VEGF and HIF-1α protein levels in all patients were not age-related and not related to the pack-yr smoking history; and 4) the reduced HIF-1α protein expression was seen in lung endothelial cells and alveolar septal cells by immunohistochemistry.
In conclusion, reduced expression of HIF-1α protein in severe COPD is consistent with the concept of a lung structure maintenance programme which is impaired on a molecular level.
- Chronic obstructive pulmonary disease
- emphysema
- hypoxia inducible factor-1α
- vascular endothelial growth factor
Chronic obstructive pulmonary disease (COPD) is a major and increasingly recognised global health problem and emphysematous lung tissue destruction accounts for a large component of the pathogenesis and morbidity of patients with this disease 1, 2. Although chronic inflammation has been identified as an important finding and documented by histological investigation of lung tissue 3–5, the cellular and molecular details of lung tissue destruction are not completely understood 6, 7. Inflammatory mediators, proteases and oxidants are released by activated cells and are thought to attack the alveolar septal structure and jeopardise the adult lung structure maintenance programme 7, 8. Vascular endothelial growth factor (VEGF) has been suggested as an integral part of such a lung structure maintenance programme 9, and the expression of VEGF and VEGF receptor (VEGFR)2 (KDR) proteins has been shown to be decreased in human lung tissue and airway samples from patients with severe COPD/emphysema 10, 11. This decrease of lung tissue VEGF gene and protein expression is unexplained, in particular, in view of the presence of inflammation and considering that patients with severe COPD are frequently hypoxic. We undertook the present study in order to investigate a mechanism which could explain the decreased VEGF protein expression in emphysematous lungs. Although the transcription factor hypoxia inducible factor (HIF)-1α is not the only factor involved in the control of VEGF gene transcription 12, 13 by binding to the hypoxia response element (HRE) 14 of the VEGF gene promoter, HIF-1α-induced VEGF expression is certainly considered a major and important mechanism of VEGF transcriptional control 15. Recently it has been recognised that hypoxia and inflammation interact on the level of HIF-1α dependent target gene expression 16, but also that there are non-hypoxic pathways of HIF-1α induction 14, 17. Because inflammation and HIF-1α overlap, it is surprising that the expression of the lung structure maintenance factor VEGF is dramatically reduced in the lungs of patients with endstage COPD 10, 11, 18.
Given the potential importance of VEGF in the maintenance of lung vascular endothelial health 19, and the finding of decreased VEGF gene expression in human emphysema 10 and animal models of emphysema 20, 21, we find it necessary to investigate the mechanisms of suppressed lung tissue VEGF gene transcription and propose impaired HIF-1α protein stability as a mechanism. In support of this hypothesis is the finding of increased p53 expression in the lungs from emphysema patients 22, as p53 regulates HIF-1α expression 23, 24. Because the transcription factor HIF-1α controls the expression of a large number of genes, a loss of HIF-1α in emphysematous lungs would have far reaching consequences, beyond VEGF gene expression, for cell growth and cell metabolism 25.
We investigated the expression of VEGF, HIF-1α, histone deacetylase (HDAC)2 and p53 proteins in samples from patients with COPD/emphysema and report a decreased expression of HIF-1α protein, which may relate to the severity of emphysema.
MATERIALS AND METHODS
Patients
We obtained lungs from 26 patients who were undergoing single or bilateral lung explantation (n = 10), lung volume reduction surgery (n = 3), lobectomy or wedge resection (n = 11) and biopsy (n = 2) for the diagnosis of lung cancer. Table 1 shows the summarised patient characteristics and online supplementary table 1 reports information of all of the patients. Samples were obtained from the National Institutes of Health Lung Tissue Repository (Bethesda, MD, USA) and from the explanted lung tissue bank of the University of Colorado Health Sciences Center Lung Transplant Program (M. Zamora). The diagnosis of emphysema was made by the Lung Tissue Repository Consortium (LTRC) pathologist (C.D. Cool) based on histological examination. The diagnosis was also supported by chest computed tomography (CT) scan data. Patients one to seven are nonsmokers, although very mild centrilobular emphysema was detected by histopathological diagnosis (except in patients one and seven), and there was no evidence of COPD. The cause or causes of the mild emphysematous changes are unknown. Patients eight to 26 are smokers, patient 10 is a current smoker and the other patients, except patient 17 (smoking history was unavailable), are former smokers. We divided the patients into three groups: no COPD (patients one to seven), mild COPD (patients eight to 13) and severe COPD (patients 14 to 26) according to the Global Initiative for Chronic Obstructive Lung Disease classification (table 1 and online supplemental table 2). The patients with severe COPD were younger than the patients with mild COPD (59.5±1.6 yrs versus 69.8±3.6 yrs; p<0.05). None of the patients' lungs showed histological findings consistent with severe pulmonary hypertension. All lung tissue samples were maintained at -80°C until processing. All of the patients gave informed consent to take part in the study and have their tissue banked in the LTRC repository. The study was approved by the Committee on Human Research (University of Colorado Health Science Center, Aurora, CO, USA).
Because we wished to validate the first data set obtained from the analysis of tissue from patients one to six and eight to 19 by subsequently analysing additional patient samples (patients seven and 20 to 26) we displayed the Western blot protein expression data separately; the first cohort data is in figure 1 and the online supplementary figures 1 and 2, the second cohort data is documented in the online supplementary figures 1 and 2.

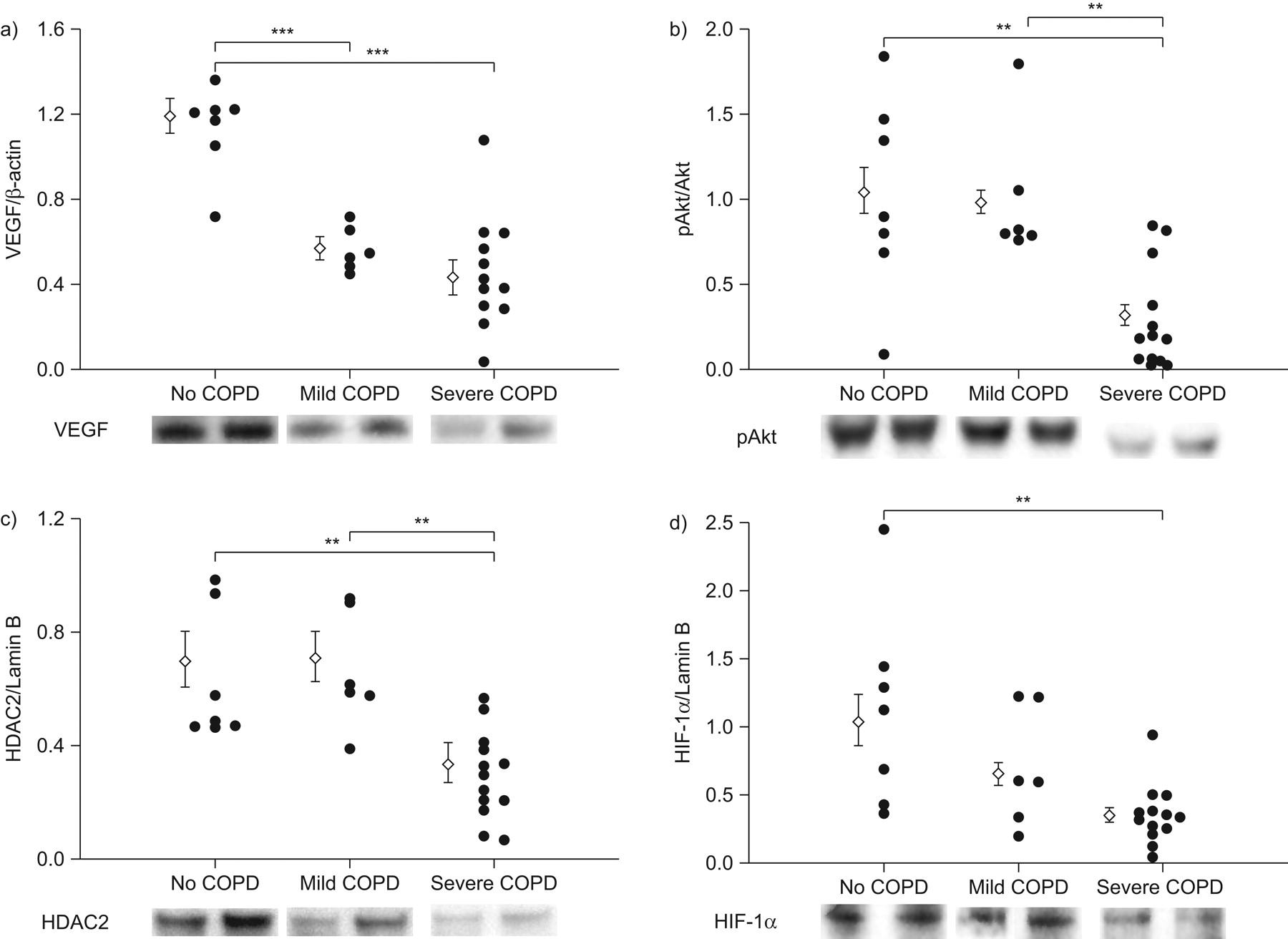
Western blot analysis of cytoplasmic and nuclear proteins. Individual densitometric data and representative protein bands are shown. Vascular endothelial growth factor (VEGF) expression was related to β-actin protein and phosphorylated Akt (pAkt) was expressed relative to the Akt protein (a and b, respectively). Histone deacetylase 2 (HDAC2) and hypoxia inducible factor (HIF)-1α expression was related to lamin B (c and d, respectively). a–d) Compared with samples from patients with normal lung function, a significant reduction in the protein expression was seen in the samples from patients with severe chronic obstructive pulmonary disease (COPD), and significant reduction of expression was seen in samples from patients with mild COPD (a). b, c) Compared with samples from patients with mild COPD, significant signal reduction was observed in the samples from patients with severe COPD. Data are expressed as mean±sem. **: p<0.01; ***: p<0.001.

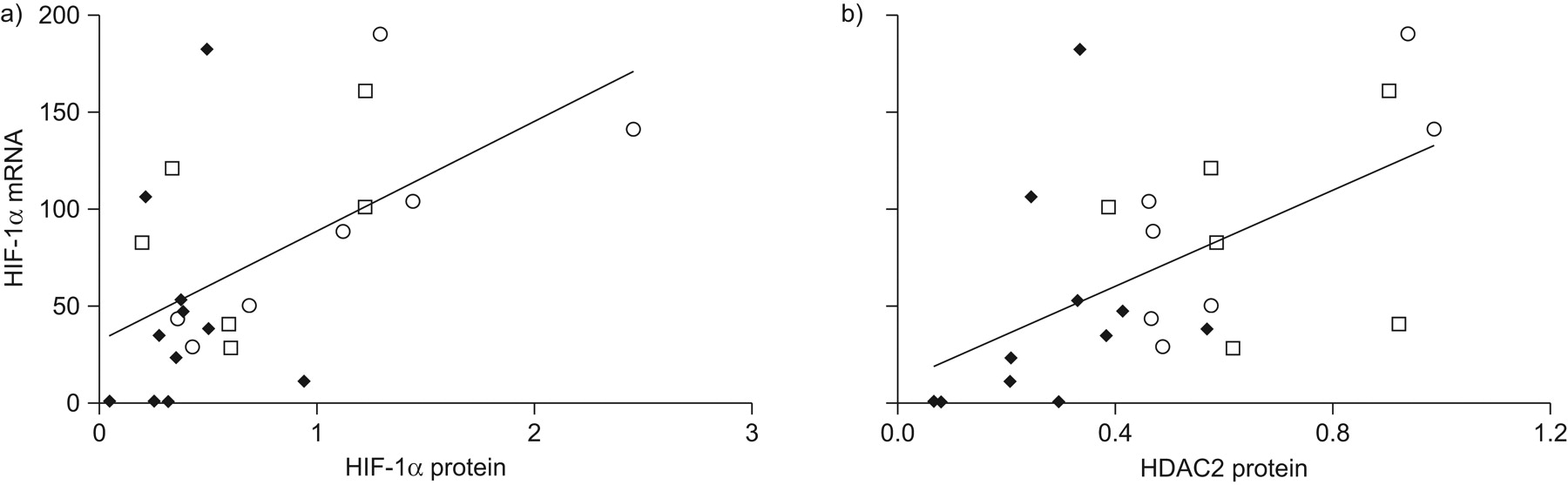
Relationship between a) histone deacetylase 2 (HDAC2) and hypoxia inducible factor (HIF)-1α proteins (R2 = 0.34, p<0.01); b) HIF-1α protein and vascular endothelial growth factor (VEGF) mRNA (R2 = 0.59, p<0.001); and c) HIF-1α protein and glucose transporter 1 (GLUT1) mRNA (R2 = 0.40, p<0.001). Both HIF-1α and HDAC2 proteins showed a strong correlation in all patients (a). A strong correlation was also seen between HIF-1α protein and the downstream gene of VEGF/GLUT-1 genes in all patients (b and c). ○: samples from patients with normal lung function; □: samples from patients with mild chronic obstructive pulmonary disease (COPD); ♦: samples from patients with severe COPD.
Chemicals
Chemicals and materials were obtained from the following sources: RNA later® ice kit frozen tissue transition solution from Ambion Inc. (Austin, TX, USA); high capacity cDNA Reverse Transcription kit from Applied Biosystems Inc. (Foster City, CA, USA); ECL system (Western Lightening® and Western Lightening® plus-ECL) from PerkinElmer (Waltham, MA, USA); cDNA reverse transcription kit and Power SYBR® Green PCR master mix from Applied Biosystems; NE-PER® Nuclear and Cytoplasmic Extraction Reagents from Thermo Scientific (Rockford, IL, USA); 4–12% Bis-Tris Nupage gels and MES-SDS running buffer from Invitrogen (Carlsbad, CA, USA); polyvinylidene difluoride membranes from Bio-Rad Laboratories (Richmond, CA, USA); protease inhibitor cocktail tablets from Roche Applied Science (Indianapolis, IN, USA); positive control of HIF-1α protein, rabbit anti-VEGF polyclonal antibody, mouse anti-Akt monoclonal antibody, rabbit anti-phospho Akt (pAkt) polyclonal antibody, mouse anti-HIF-1α monoclonal antibody, rabbit anti-HDAC2 polyclonal antibody, goat anti-lamin B polyclonal antibody, rabbit anti-factor inhibition of HIF-1α -1 (FIH-1) polyclonal antibody, mouse anti-p53 monoclonal antibody and horseradish peroxidase-conjugated goat anti-mouse and rabbit and donkey anti-goat immunoglobulinG from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA); mouse anti-heat shock protein (Hsp)90 monoclonal antibody from Stressgen bioreagents (Ann Arbor, MI, USA); Surveyor™ IC human/mouse total HIF-1α immunoassay from R&D systems, Inc (McKinley Place, MN, USA); Vectastain® Elite ABC-Peroxidase Kits Universal from Vector Laboratories (Burlingame, CA, USA); liquid diaminobenzidine substrate chromogen system was from Dako North America Inc. (Carpinteria, CA, USA). All other chemicals were purchased from Sigma (St. Louis, MO, USA).
Real-time RT-PCR analysis of lung tissue
The sequence of the forward and reverse primers is shown in online supplementary table 2. Additional details of the methods are provided in the online supplementary material.
Western blot analysis of lung tissue
To extract nuclear and cytoplasmic proteins, stored fresh frozen lung tissue samples from patients were properly manipulated on dried ice. Additional details of the methods are provided in the online supplementary material.
Total HIF-1 α protein quantification of lung tissue
Total HIF-1α protein quantification by ELISA method was performed using the Surveyor™ IC human/mouse total HIF-1α immunoassay kit (R&D Systems, Inc.) and the manufacturer's instructions with some modifications.
Immunohistochemistry of HIF-1α
Formalin fixed resected lung tissue from patients with or without COPD was examined. Five-micron random lung tissue sections were mounted onto Superfrost slides (VWR International, West Chester, PA, USA). Immunohistochemistry of HIF-1α was then performed as previously described 26. Additional details of the methods are provided in the online supplementary material.
Statistical analysis
All data are expressed as mean±sem. Clinical data were evaluated for significance by ANOVA and the Scheffe test, and differences in other data were analysed by the Kruskall–Wallis test. Correlations were analysed by the Pearson correlation coefficient. Significance was determined at p<0.05 (two-tailed test).
RESULTS
Patient characteristics
Summarised patient characteristics are shown in table 1. All of the patient data, including the CT scan data and the pathological diagnosis, with the exception of patients seven and 20 to 26, were extracted from the LTRC database (online supplementary table 1). Some, albeit mild degree of emphysema was detected histologically in all of the lung tissue samples from patients with normal spirometric values, with the exception of patients one and seven. 10 of the 26 tissue samples were tumour-free tissues from patients diagnosed with lung cancer and three patients had a history of oral steroid use. All of the patients had centrilobular emphysema and all of the patients without a clinical diagnosis of COPD (providing tissue samples one to seven) were nonsmokers. The pack-yr cigarette smoking history of the COPD patients varied between 10 and 120; for one of the COPD patients the smoking history was unknown. The degree of lung function impairment ranged from mild to very severe. The patients are listed in the online supplementary table 1 in the order of their % predicted forced expiratory volume in 1 s (FEV1).
Lung tissue expression of cytoplasmic and nuclear proteins
Western blot protein analysis was performed for VEGF and pAkt. The protein expression was referenced to β-actin and Akt, respectively. The data confirm previous findings of decreased VEGF protein expression in random lung tissue samples from patients with severe COPD/emphysema 10 and, in addition, show reduction in lung tissue pAkt protein expression (fig. 1a and b, and online supplementary fig. 1a), which has not been previously reported. In the same lung tissue samples, nuclear proteins were assessed and referenced to the house keeping protein lamin B (fig. 1c and d and online supplementary fig. 1b). Individual data and representative protein expression by Western blot for VEGF, pAkt, HIF-1α and HDAC2 in the categories no COPD, mild COPD and severe COPD are shown in figure 1. As can be seen, the expression of the HDAC2 protein was reduced in most of the tissue samples from patients with severe COPD/emphysema confirming earlier reports (fig. 1c and online supplementary fig. 1b) 27. Of interest, HIF-1α protein also showed a reduction in expression in the samples from some patients with less severe COPD/emphysema (fig. 1d and online supplementary fig. 1b). To confirm that indeed HIF-1α protein expression was reduced in the lung tissues, we used an ELISA and found that HIF-1α protein expression by ELISA was significantly decreased (data not shown) in the lungs from patients with severe COPD (p<0.005 versus no COPD). Likewise, again in the same tissue samples, there was a significant increase in the expression of p53 in lungs from COPD/emphysema patients (p<0.05) by densitometric analysis when severe COPD patient samples were compared with samples from patients that had normal lung function. There was no difference in the tissue expression of the prolyl hydroxylase-2 (PHD-2) protein and FIH-1 and also no statistically significant difference in the expression of the chaperone involved in HIF-1α expression control, Hsp90 (online supplementary fig. 2).
HIF-1α protein is correlated with HDAC2 protein
We found that HIF-1α protein expression related to HDAC2 protein expression (R2 = 0.34, p<0.01) (fig. 2a) and HIF-1α protein expression was also strongly correlated with the target genes VEGF (R2 = 0.59, p<0.001) (fig. 2b) and glucose transporter 1 (R2 = 0.40, p<0.001) (fig. 2c).
HIF-1α protein expression in lung tissue and severity of COPD/emphysema
All individual FEV1 % pred data that we could obtain (n = 26) are shown in figure 3a. When all of the tissue samples from patients with or without COPD were considered, a significant correlation between HIF-1α and FEV1 was found (R2 = 0.32, p<0.005) (fig. 3b). When the lung tissue HIF-1α protein expression was plotted against pack-yrs of cigarettes smoked, we did not find a correlation between those two variables (online supplementary fig. 3). Since there was a significant age difference between patients with mild and severe COPD (table 1), we analysed the relationship between age and VEGF and HIF-1α protein levels; there was no age-dependent relationship (R2 = 0.018, p = 0.51 and R2 = 0.0008, p = 0.89, respectively).

A significant positive relationship between forced expiratory volume in 1 s (FEV1) % predicted and hypoxia inducible factor (HIF)-1α protein was shown (R2 = 0.32; p<0.005). ○: samples from patients with normal lung function; □: samples from patients with mild chronic obstructive pulmonary disease (COPD); ♦: samples from patients with severe COPD. Data are expressed as mean±sem.
Figure 4 shows the immunohistological localisation of HIF-1α protein in a tissue section from a normal lung (fig. 4a and b) and one sample from a lung obtained from a patient with severe COPD (fig. 4c and d). The lung from the severe COPD patient demonstrates less expression of pulmonary artery endothelial cell HIF-1α protein expression. The HIF-1α expression by alveolar septal cells is also reduced.

Hypoxia inducible factor (HIF)-1α protein localisation analysis by immunohistochemistry. a, b) Normal human lung and c, d) a lung tissue section from a patient with severe chronic obstructive pulmonary disease (COPD) were examined. a, c) Arrows show the endothelial cell layer. b, d) Arrows show HIF-1α positive alveolar septal cells. Reduced HIF-1α positivity was seen in both pulmonary endothelial and lung septal cells. Similar results were obtained when additional samples from patients with COPD were examined (not shown). Scale bars = 50 μm. e) The ratio of HIF-1α positive cells and total length of alveolar perimeter (TLAP) in lung sections from a patient with no COPD or severe COPD. Data are expressed as mean±se. *: p<0.05 versus no COPD.
Gene expression changes in COPD/emphysema lung tissue
When we related the tissue expression of HIF-1α mRNA/β-actin mRNA to the corresponding HIF-1α protein expression, we found a correlation between lung tissue HIF-1α mRNA and protein expression (R2 = 0.30; p<0.01) (fig. 5a), indicating that in COPD/emphysema lung tissue both HIF-1α transcription and protein stability and degradation had been affected. Surprisingly, a significant positive correlation between HDAC2 protein and HIF-1α gene expression was also found (R2 = 0.31; p<0.01) (fig. 5b).

a) Relationship between hypoxia inducible factor (HIF)-1α gene and protein expression (R2 = 0.30; p<0.01) . b) Relationship between HIF-1α gene and histone deacetylase 2 (HDAC2) protein (R2 = 0.31; p<0.01). A significant correlation was seen between the HIF-1α gene and these two proteins. ○: samples from patients with normal lung function; □: samples from patients with mild chronic obstructive pulmonary disease (COPD); ♦: samples from patients with severe COPD.
DISCUSSION
Modern views of the pathobiology of emphysema include inflammatory and immune mechanisms 28, 29 and the concept of a breakdown of a molecular lung structure maintenance programme 7, 30. We describe for the first time the decreased expression in lung tissues from patients with COPD/emphysema of a nuclear protein, HIF-1α , which is critically involved in the control of gene expression. We also confirm the previously reported decreased expression of another nuclear protein, HDAC2 27. The reduced expression of HIF-1α in emphysematous lung tissues is a surprising finding in view of the known induction/stabilisation of this protein by hypoxia and inflammatory mechanisms 31. HIF-1α expression is increased in hypoxaemic and inflamed tissues, and in many tumour tissues HIF-1α is known to promote tumour angiogenesis 32. In contrast, no diseases have so far been described that are characterised by a decrease of tissue levels of the HIF-1α protein, with the exception of acute ischaemia and reperfusion of the myocardium 33, 34 and the respiratory distress syndrome in pre-term lambs 35. Decreased or impaired activity of this transcription factor has profound consequences for the homeostatic control at the cellular and mitochondrial level and will probably affect apoptosis and cellular senescence which characterise pulmonary emphysema 20, 22.
In general, HIF-1α protein degradation is mediated by PHD, the von Hippel-Lindau/Elongin-C/B E3 ubiquitin ligase complex and the proteasome 15. The HIF-1α transactivation domain is also regulated by the binding of FIH-1 15. All of these control mechanisms are on the levels of protein degradation and/or post-transcriptional regulation.
Histone deacetylases are of equal fundamental importance for transcriptional control. The histone deacetylase family of chromatin-modifying enzymes participates in post-translational modifications involved in epigenetic regulation. Other studies 10, 11 have previously reported a significant decrease in the expression of VEGF and VEGFR2 (KDR) mRNA and protein in the lung tissue from patients with severe COPD/emphysema 10, and because a similar VEGF expression decrease had been reported in the lungs from animals exposed to lethal hyperoxia 36, we had hypothesised that overwhelming oxidant stress may destroy the transcriptional machinery which regulates VEGF expression. Because tissue samples from patients with lung cancer were included in our study, we considered the possibility of an enhanced expression of HIF-1α and VEGF in such patients. However, the samples were from tumour-free lung lobes and enhanced HIF-1α or VEGF expression was not found. For example, patient 11 had lung cancer but this patient's HIF-1α protein expression was the lowest in mild COPD tissue (online supplementary fig 1b). Given the consensus of the importance of oxidative stress in cigarette smoking-related lung tissue destruction 6 and the data which show that HDAC2 expression is impaired by oxidative stress 27, 37 and that p53 protein is upregulated in response to cellular stress, it is difficult to determine a single initial or primary event in the development of emphysema related to tobacco smoke exposure. In figure 6, we propose mechanistic interactions between several of the pathobiologically important components, which may, in part, also play a role in emphysema which develops in nonsmokers.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic diagram representing the molecular alterations in the emphysematous lungs. Oxidative stress may inhibit histone deacetylase 2 (HDAC2) protein expression and increase p53 protein expression. The reduction of HDAC2 may upregulate p53 expression. Both upregulated p53 and reduced HDAC2 could reduce the hypoxia inducible factor (HIF)-1α protein. Insufficient HIF-1α causes impaired expression of downstream mRNA encoding vascular endothelial growth factor (VEGF) and glucose transporter 1 (GLUT1) and may impair the binding to VEGF receptor and phosphorylation of Akt, which could cause lung cell apoptosis and emphysema. Decreased expression of the VEGF receptor protein has been previously reported 10. PHD-2: prolyl hydroxylase-2; Hsp90: heat shock protein 90.
Because HIF-1α protein is a major transcriptional controller of VEGF expression, and oxidative stress has been widely documented in human emphysematous lungs 38 and animal models of emphysema 39, 40, we reason that oxidative stress, directly or indirectly affects HIF-1α protein expression. Of associated interest is the finding that in COPD lungs both VEGF 10 and HIF-1α protein expression is reduced in pulmonary arterial endothelial cells and pulmonary septal cells (fig. 4). Recently, Charron et al. 41 reported in A549 cells that hypoxia-induced HIF-1α regulates HDAC2 transcription. Whether HIF-1α controls HDAC expression in lungs from COPD patients remains unclear.
The analysis of human tissue samples confirmed the reduction in the expression of VEGF protein and now also shows a reduction in the expression of the downstream cellular growth regulating protein pAkt in tissues from patients with severe COPD (fig. 1a and b and online supplementary fig. 1a).
The lung tissue levels of HIF-1α protein did not show a statistically significant correlation with the pack-yr smoking history (online supplementary fig. 3), however, they did correlate with the % pred FEV1 (fig. 3). In spite of the low level HIF-1α expression by Western blot (fig. 1 and online supplementary fig. 1) and ELISA (data not shown) in the majority of the lungs from COPD patients, we did attempt immunohistochemical HIF-1α localisation as illustrated in figure 4 and found a reduced expression of HIF-1α protein in pulmonary endothelial cells and pulmonary septal cells.
Upregulation of the pro-apoptotic p53 in human emphysema has been described by Tsuji et al. 22. Increased p53 expression is indeed a consequence of HDAC inhibition 42, and our own data also show increased p53 expression in severe COPD/emphysema lung tissue samples (online supplementary fig. 2). Using human pulmonary microvascular endothelial cells and lung fibroblasts we knocked down HDAC2 gene expression and observed, as a consequence, decreased HIF-1α expression 43. The significance of the increased expression of p53 in the COPD lung tissues may lie in the p53-dependent control of the expression of HIF-1α.
We have also analysed the tissue samples for expression of Hsp90, a chaperone protein which forms complexes with HDAC6 and affects HIF-1α protein stability 44, 45. Using Western blot analysis we did not find a significant reduction in the tissues from patients with COPD/emphysema compared with tissues from patients with normal lung function (online supplementary fig. 2a and b).
It may be possible to characterise human emphysema in smokers also as a “HIF-1α deficiency” problem. HIF-1α protein abundance may be reduced as a consequence of a combination of several factors among others: increased p53 expression (fig. 6). Figure 6 illustrates our attempt to connect signalling events both upstream and downstream from VEGF. Whereas Kasahara et al. 10 reported decreased expression of VEGFR2 (KDR) in human emphysema lungs, we now also report on decreased Akt phosphorylation and can connect decreased HIF-1α expression and Akt phosphorylation. Certainly Akt phosphorylation is determined by additional factors. What little residual HIF-1α protein remains in the COPD/emphysema lung may bind ineffectively to the HRE of target gene promoters as elegantly demonstrated by Breit et al. 46 and Ruchko et al. 47 for the VEGF transcriptional impairment under conditions of oxidative stress. Our study is limited by the relatively small number of patient samples and the lack of available data relating to the arterial oxygen tension of the patients. It is also possible that there may be regional expression differences of the genes and proteins between upper and lower lung lobes, such that potentially site-specific expression differences could not been examined. As is evident, causal and mechanistic data cannot be obtained from single time-point tissue samples and must be complemented by animal and cell model data 43.
In conclusion, we demonstrated decreased HIF-1α protein expressions in emphysematous lungs. The mechanisms whereby HIF-1α expression is reduced may include p53-dependent and p53-independed pathways as proposed in a recent publication 48. In a greater context, we are now able to appreciate the problem of emphysematous tissue destruction also as an impairment of nuclear-transcriptional events. Given the postulated autocrine role of VEGF in controlling lung microvessel endothelial cell survival 10, our data point towards a double jeopardy: VEGF transcription is limited by HIF-1α protein stability and the action of VEGF in the severely emphysematous lungs 20 is limited by a decreased VEGF receptor expression.
Acknowledgments
We would like to thank V. Kraskauskasiene (Victoria Johnson Center, Virginia Commonwealth University, Richmond, VA, USA) for expert technical assistance.
Footnotes
↵This article has supplementary material available from www.erj.ersjournals.com
Support Statement
Supported by the National Institute of Health Lung Division Lung Tissue Repository Consortium grant support mechanism (grant number N01-HR-46160-3; N.F. Voelkel). Supported by the Victoria Johnson Center for Obstructive Lung Diseases (Richmond, VA, USA).
Statement of Interest
None declared.
- Received February 10, 2010.
- Accepted June 4, 2010.
- ©2011 ERS
REFERENCES










