





Abstract
Inflammation is a main pathogenetic factor in the development and progression of chronic obstructive pulmonary disease (COPD). Recently, it has become clear that not only the innate, but also the specific immune response plays a role. A striking finding, in particular in lungs of patients with severe COPD, often with a predominant emphysema phenotype, is the presence of B-cell follicles. As seen in other tissues, these follicles are the result of lymphoid neogenesis. The finding of oligoclonality in B-cell follicles in COPD suggests that they play a role in local antigen specific immune responses. To date, it is not known which antigens may be involved; microbial antigens, cigarette smoke-derived antigens and antigens from extracellular matrix breakdown products have been suggested. Consequently, the pathogenetic role of this follicular B-cell response is not yet clear. It might be protective against microbial colonisation and infection of the lower respiratory tract and, therefore, beneficial, or it could be of a more harmful (autoimmune) nature, directed against lung tissue components. It is necessary to determine the specific antigen(s) and to explore the exact role of the COPD related B-cell response in order to include modulation of this response and develop therapeutic options.
Chronic obstructive pulmonary disease (COPD) is a chronic inflammatory disorder in different compartments of the lung, including a mixture of small airway disease (obstructive bronchiolitis) and parenchymal destruction (emphysema). COPD is associated with a relentlessly progressive course and increasing morbidity, disability and mortality worldwide 1. Smoking is known to be the major aetiological factor, yet the exact pathogenetic mechanisms have not yet been elucidated. Smoking cessation is the only effective treatment, but it only partially attenuates further loss of lung function. In those COPD patients who have fully ceased smoking, pulmonary inflammation persists 2–4. The mechanism that propagates this inflammation is incompletely understood.
Since COPD is characterised by chronic airflow limitation that is not fully reversible and usually progressive, the Global Initiative for Chronic Obstructive Lung Disease (GOLD) has introduced a five-stage classification for the severity of COPD based on measurements of airflow limitation during forced expiration, as measured by forced expiratory volume in 1 s 5. The progression of COPD is associated with infiltration of the wall of small airways by innate and adaptive inflammatory immune cells. The inflammatory cells that were initially identified as being the most important in the pathogenesis of COPD were neutrophilic granulocytes and macrophages, which both belong to the innate immune system. The importance of neutrophils and macrophages has been confirmed by animal studies showing the necessity of both these cells for the induction of experimental emphysema by cigarette smoke (CS) 6, 7. It later became clear that the specific immune system also plays a role; in particular CD8 positive T-lymphocytes were associated with smoking and subsequent risk for COPD 8, 9. Recently, B-lymphocytes organised into peribronchiolar lymphoid follicles have been associated with severe COPD 10 and, in addition, in COPD such follicles were also found in the parenchyma 11, whereas B-cells were increased in large airways 12. As the antigen inducing this follicular B-cell response is as yet unknown, it has yet to be decided whether and to what extent this response is of beneficial or harmful nature. This review aims to shed light on the nature of this response, the possible aetiological factors and to present our views on the possible role in the pathogenesis and progression of COPD.
LYMPHOID FOLLICLES IN COPD
Whereas one of the first reports on the presence of B-cell follicles in COPD dates from 1992 13, quite recently attention has been redrawn to a possible role of B-cells. An increase of small airways containing B-cell follicles was found in patients with severe COPD compared to normal subjects and patients with mild-to-moderate COPD. Furthermore, the progression of COPD from GOLD stage 0 to GOLD stage 4 was indeed clearly associated with the number of small airways containing these lymphoid follicles 10, 14. Furthermore, the presence of B-cell follicles was demonstrated not only in relation to small airways, but also in the lung parenchyma of COPD patients (fig. 1⇓) and mice chronically exposed to CS 11. An increase of individual B-cells in COPD was also observed, in particular in bronchial biopsy studies of large airways and, in addition, nonfollicular B-cell aggregates were identified 12, 15.

Lymphoid follicles in chronic obstructive pulmonary disease. a) CD20: overview of emphysematous lung tissue with B-cell follicles apposing a small airway (arrows) and a small B-cell aggregate in the parenchyma (arrowhead). b) CD20 staining of all B-cells. c) CD3 staining of apposing T-cells. d) CD21 (complement C3d-receptor) showing delicate staining of follicular dendritic cells. e) CD35 (complement C3b-receptor) showing more intense staining of follicular dendritic cells and weak staining of B-cells. f) CD83 staining of mature myeloid dendritic cells, showing subepithelial location (arrows), as well as presence in the rim (T-cell zone) of a lymphoid follicle (arrowhead). a) Scale bars = 1,000 μm, b–f) scale bars = 200 μm.
It is important to note that the original publication demonstrating lymphoid follicles in patients with severe and very severe COPD 10 could encompass a selection bias, since all lung specimens of GOLD stage 3 and 4 COPD were obtained from lung volume reduction surgery specimens of patients with severe emphysema enrolled in the National Emphysema Treatment Trial (NETT). However, in the study of van der Strate et al. 11, the majority of the human lung tissue samples (six out of eight patients with very severe COPD) were obtained at lung transplantation (explant lungs). Nevertheless, more studies are needed to assess the role of lymphoid follicles in different COPD disease stages and phenotypes, including severe COPD with predominant bronchiolitis.
With respect to B-cell function and development, a distinction should be made between B-cell aggregates without a specific functional architecture and B-cells organised in (primary or secondary) follicles; this is also reflected in their supposed functional role. Lymphoid aggregates consist of mature, memory B-cells and/or T-cells without functional organisation, thus mainly available for local priming and activation. The functional organisation (see below) of primary and secondary lymphoid follicles (the latter containing germinal centres) consists of a specific arrangement of memory and naїve B-cells, T-cells, dendritic cells (DCs) and follicular dendritic cells (FDC), which allows for B- and T-cell priming, clonal expansion, antigen retention (mostly as immune complexes), somatic hypermutation, affinity maturation and immunoglobulin (Ig) class switching 16–18. However, it should be kept in mind that over time the distinction is not always clear. When local circumstances and factors allow, lymphoid follicles can develop out of any lymphoid aggregate. Importantly, only the micro-anatomic organisation of a lymphoid follicle, with the ability of germinal centre development, allows for its specific functions 16–18.
In COPD, the follicles in the lung 10, 11, 14 consist of large aggregates of B-cells that are surrounded by lower numbers of mainly CD4 (80–90%) and some CD8-positive T-cells. These aggregates can be considered lymphoid follicles, since FDC were present in these aggregates, expressing CD21 and CD35 (fig. 1⇑). The presence of FDC, involved in affinity maturation and isotype switching of B-cells, suggests antigen-driven proliferation. The B-cells were mainly IgM-positive and IgD-negative, which suggests that these B-cells may have been activated to some extent. Moreover, a predominant part of the infiltrate appeared to be CD27-positive, a marker for memory B-cells 19. Interestingly, CD138-positive (plasma) cells were detectable in the direct vicinity of these follicles. Furthermore, CD40 and CD40 ligand, important co-stimulatory molecules, were expressed in these follicles. The Ki-67 antigen was detected on small central clusters of B-cells in the follicles, suggesting an early germinal centre reaction 17.
Most importantly, B-cells isolated from the B-cell follicles have been demonstrated to be oligoclonal in nature 11, suggesting antigen-specific induction of the B-cell follicles. In this study, 10 lymphoid follicles were isolated by laser microdissection from the lung tissue of eight patients with COPD 11. Sequence analysis of the Ig rearrangements revealed the presence of 12 different B-cell contigs with one or more sequence variation in 10 of the contigs. In seven out of the eight patients, clonal B-cell populations were observed, whereas in one patient only unrelated sequences were observed. The seven patients with related Ig sequences demonstrated the presence of ongoing mutations. The latter indicates oligoclonal B-cell proliferation in response to stimulation with as yet unknown antigen(s).
With respect to B-cell follicles in the lung, some authors regarded these as part of the mucosal immune system and thus as so-called bronchus associated lymphoid tissue. However, in most normal lungs no lymphoid tissue is observed at all 20. Lymphoid follicles have been observed in the human lung in (subclinical or clinical) lung disease, either associated with airways or with parenchyma. Consequently it seems most logical to regard this as so-called lymphoid neogenesis and, thus, as an ectopic lymphoid tissue 16, 21. Such lymphoid tissues are also called tertiary lymphoid organs (TLOs), as opposed to primary (bone marrow and thymus) and secondary lymphoid organs (lymph nodes, Peyer’s patches and spleen).
The term lymphoid neogenesis refers to the development of organised lymphoid structures which resemble secondary lymphoid organs in tissues that are targeted by chronic inflammatory processes, such as infection and autoimmunity 16, 22. The purpose of immune responses is primarily the eradication of pathogens. However, certain antigens are difficult to eradicate and result in sustained immune responses leading to chronic inflammation. In tissues that harbour the target antigens of chronic adaptive immune responses, infiltration occurs by macrophages, DCs, T-cells, B-cells and plasma cells. These cells frequently organise themselves at an anatomic and functional level as in secondary lymphoid organs, leading to the formation of B-cell follicles and T-cell areas 23. This lymphoid neogenesis, also called TLO formation, is a dynamic process starting with sparse lymphocytic infiltrates that evolve into aggregates and eventually organise in B-cell follicles with germinal centres and distinct T-cell areas containing DCs and high endothelial venules (fig. 2⇓). High endothelial venules regulate the extravasation of naïve B- and T-cells. Thus, there are remarkable similarities between the structure of secondary lymphoid organs and TLOs, including the compartmentalisation in distinct B-cell areas (follicles) and T-cell areas (paracortex). However, in contrast to lymph nodes, TLOs are not supplied by afferent lymph vessels and are not encapsulated.
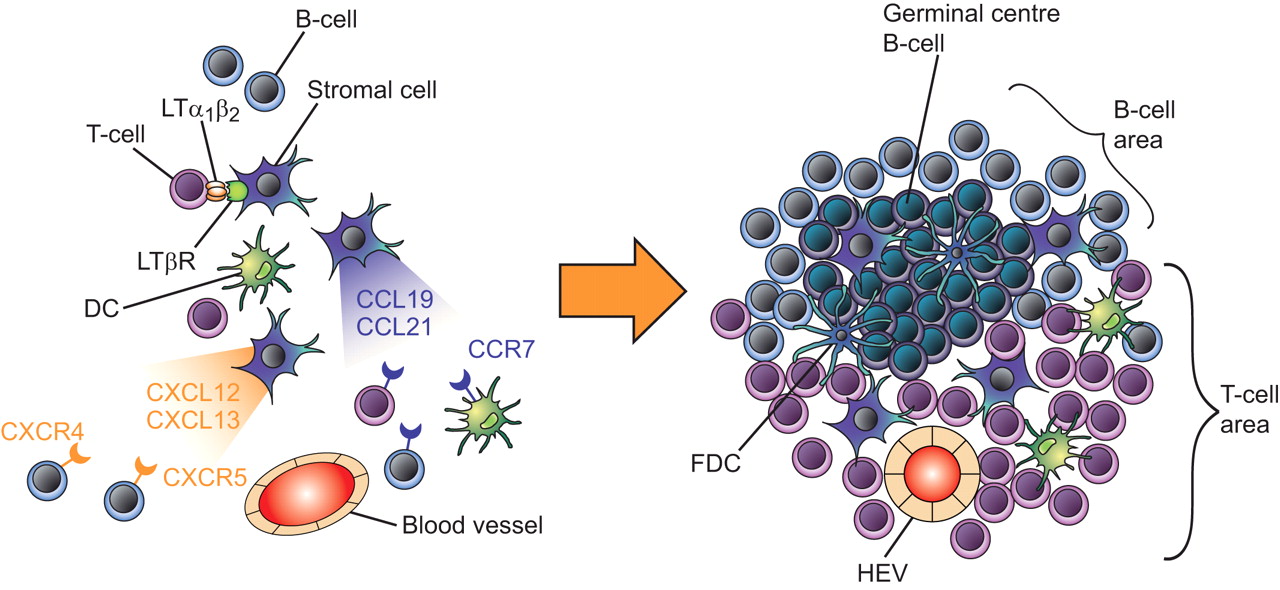
Lymphoid neogenesis in chronic inflammation. a) In chronic inflammation, activated lymphocytes expressing lymphotoxin-α-β heterotrimer (LTα1β2) interact with the lymphotoxin (LT)β receptor on neighbouring stromal cells. The stromal cell stimulation induces the expression of lymphoid chemokines (CC-chemokine ligands (CCL)19, CCL21, CXC-chemokine ligands (CXCL)12 and CXCL13) along with adhesion molecules allowing the recruitment of naїve lymphocytes and dendritic cells (DCs). b) Prolonged inflammatory stimulation will lead to the organisation of lymphoid aggregates into lymphoid follicles. Germinal centres may form within the B-cell zone with highly specialised follicular dendritic cells (FDC). High endothelial venules (HEV) expressing lymphoid chemokines and adhesion molecules ensure lymphocyte and DC recruitment.
LYMPHOID NEOGENESIS IN MURINE MODELS OF COPD
In vivo animal models can help to unravel the cellular and molecular mechanisms underlying the pathogenesis of COPD in general and of lymphoid neogenesis in particular 24–26. Mice represent the most favoured animal species with regard to the study of the immunological mechanisms of diseases, as several hundreds of inbred strains and specialised stocks (e.g. mutants) are available and since they offer the opportunity to manipulate gene expression (e.g. transgenic and knock-out mice). Lymphoid follicles have been described not only in CS-based murine models of COPD, but also in pathogen-based models of COPD and in transgenic mice over-expressing pro-inflammatory cytokines in the bronchial or alveolar epithelium.
CS-induced lymphoid follicles
When the time-course of CS-induced pulmonary inflammation is determined in wild-type mice, a progressive accumulation of innate inflammatory cells (i.e. neutrophils, macrophages and DCs) in the airways and lung parenchyma is demonstrated from day 3 onwards. In addition to the persistent accumulation of innate inflammatory cells upon prolonged exposure to CS, a clear and progressive infiltration of adaptive immune cells (i.e. B-cells and T lymphocytes, both CD4+ and CD8+ T-cells) has been observed in bronchoalveolar lavage fluid and lungs of mice exposed to CS for 3–6 months 11, 27. Inflammatory infiltrates, with a predominantly lymphoid character, were found to be present around bronchioles and in the lung parenchyma of mice after chronic CS exposure, similar to the lymphoid follicles detected in lung sections of patients with COPD. Lymphoid follicles consisting of B-cells and FDC with adjacent T-cells were detected in mice that had developed pulmonary inflammation after smoking for 6 months 11. The increase in the number and the size of B-cell follicles was progressive with time and correlated with the (progressive) enlargement of alveolar airspaces, as evidenced by an increase in mean linear intercept. The development of emphysema in CS-exposed mice has been shown to be strain dependent. Although lymphoid follicles have been described in several strains of CS-exposed mice (e.g. BALB/c, C57BL/6 and C3H) 27–29, a formal head-to-head comparison of lymphoid neogenesis in different murine strains has not yet been performed. The number and size of peribronchial lymphoid follicles was significantly attenuated in chronically CS-exposed chemokine receptor CCR5 and CCR6 knock-out mice compared with wild-type animals, indicating a prominent role of chemokines in lymphoid neogenesis 28, 30.
Pathogen and lipopolysaccharide-induced lymphoid follicles
Several bacteria, including nontypeable Haemophilus influenzae (NTHi), commonly colonise the lower airways of patients with (severe) COPD. Exposure of mice to an aerosolised NTHi lysate every 8 weeks induced infiltration of macrophages, CD8+ T-cells and B-cells around airways and blood vessels, including rounded peribronchial lymphoid aggregates 31. Lipopolysaccharide (LPS) is a major pro-inflammatory glycolipid component of the bacterial cell wall of Gram-negative bacteria (such as NTHi). Interestingly, repeated intratracheal instillation of LPS in mice resulted in persistent chronic pulmonary inflammation, characterised by peribronchial and perivascular lymphocytic aggregates (consisting of CD4+ and CD8+ T lymphocytes and CD19+ B lymphocytes) 32. These in vivo studies demonstrate that some pathological changes of COPD, especially lymphoid aggregates, can be mimicked in mice after repeated exposure to either Gram-negative bacterial lysates or a component of the Gram-negative bacterial cell wall, i.e. LPS. Finally, Moyron-Quiroz et al. 33 demonstrated that an infectious challenge with influenza virus in mice can trigger lymphoid chemokine expression and lymphoid neogenesis in the lungs, even in lymphotoxin (LT)α-deficient mice that lack secondary lymphoid organs 33.
Spontaneous development of lymphoid aggregates in lungs of transgenic mice
Transgenic mice were generated in which human interleukin (IL)-1β is conditionally (with a doxycycline-inducible system) and specifically (by the Clara cell secretory protein promoter) expressed in the lung epithelium, especially in the bronchioli 34. Induction of IL-1β expression in the lungs of adult mice caused pulmonary inflammation, distal airspace enlargement, airway wall fibrosis and lymphocytic aggregates in the airways. Human IL-6 and IL-6 receptor (IL-6R) double transgenic mice also developed mononuclear cell accumulation in peribronchovascular regions and lymphoid tissue-like structures expressing the chemokine CXCL13, which is indispensable for lymphoid organogenesis 35. In separate studies, transgenic mice over expressing tumour-necrosis-factor (TNF)-α in the lung under the surfactant protein C (SPC) promoter have been shown to produce elements of both emphysema and pulmonary fibrosis 36, 37. Detailed histological analysis of SPC/TNF-α transgenic mice showed major heterogeneous abnormalities of the lung parenchyma, including infiltration of macrophages and lymphocytes into the lungs, which was most prominent in the subpleural regions, as well as distortion of alveolar structures due to septal thickening 38. Since the pro-inflammatory cytokines IL-1β, IL-6 and TNF-α have pleiotropic effects and have been implicated in multiple inflammatory disorders, it is clear that the observed pathological abnormalities in these transgenic mice can be involved in several human diseases, including not only COPD/emphysema but also lymphocytic fibrosing alveolitis, leading to pulmonary fibrosis.
LYMPHOID NEOGENESIS IN OTHER DISEASES
Lymphoid follicles are observed in various lung diseases other than COPD. Often, a long-term fibrosing process has been going on with, in due time, development of lymphoid aggregates and lymphoid follicles. This is observed in usual interstitial pneumonia/end-stage pulmonary fibrosis and also in nonspecific interstitial pneumonia and bronchiectasis. In bronchiectasis, follicles are not only observed in the airway wall but also in the surrounding parenchyma, which often shows varying fibrosis and chronic inflammation. In most of the above conditions, stasis of cellular debris and mucus is present in distorted airspaces, in which all kinds of microorganisms are present, often with clinical or subclinical infections. Therefore, an antimicrobial origin of the specific immune response giving rise to the follicles is quite likely in these diseases, although other origins cannot be excluded.
Lymphoid interstitial pneumonia (LIP) is an interstitial lung disease characterised by an extensive proliferation of small mature B- and T-cells; in general, mainly in the alveolar interstitium. However, a form with follicular bronchiolitis is often also considered to be part of this spectrum. Development of lymphoid follicles, most with florid germinal centres, is a general feature of LIP. At present LIP is considered a pulmonary manifestation related to different autoimmune diseases; auto-antigens are also most likely underlying the immune response giving rise to the lymphoid follicles. With respect to the airways, follicles are seen in bronchiectasis as described above, but the most prominent airway manifestation of lymphoid follicles is in follicular bronchiolitis. This disease condition is quite rare, sometimes related to autoimmune disease, and often considered as part of LIP (see above). As a pathological diagnosis it has been observed in association with immunodeficiency, such as in HIV, but also as an idiopathic disease.
Lymphoid neogenesis has also been described outside the lungs in several chronically inflamed tissues from patients with different underlying diseases, mainly autoimmune and infectious diseases. Human chronic inflammatory diseases with lymphoid neogenesis encompass rheumatoid arthritis, Graves’ disease (hyperthyroidism), Hashimoto’s thyroiditis (hypothyroidism), myasthenia gravis, multiple sclerosis, Crohn’s disease, ulcerative colitis and chronic hepatitis C. The respective target tissues of the TLO formation are diverse and include the joints, the thyroid gland, the thymus, the central nervous system, the gut and the liver. In several autoimmune diseases, the self-antigen, recognised by antibodies generated in ectopic germinal centres, has been detected in lymphoid follicles (e.g. thyroglobulin and thyroperoxidase in Hashimoto’s thyroiditis) 16.
CELLULAR AND MOLECULAR MECHANISMS OF LYMPHOID (NEO)GENESIS
Similarities in development of secondary lymphoid organs versus TLOs
The organisation of lymph nodes during organogenesis is the result of a highly coordinated interplay between haematopoietic cells, mesenchymal stromal cells, adhesion molecules, cytokines and chemokines (fig. 3⇓). The key players are members of the TNF family, mainly LTα1β2 and its receptor LTβR 39, 40. Binding of membrane-bound LTα1β2, expressed by haematopoietic inducer cells to LTβR on the surface of stromal organiser cells, induces the expression of adhesion molecules, including intercellular adhesion molecule-1, vascular cell-adhesion molecule-1, mucosal addressin cell-adhesion molecule-1 and peripheral node addressin. The interaction between haematopoietic inducer cells and stromal cells via the LTα1β2–LTβR pathway also induces the expression of homeostatic lymphoid chemokines, such as CC-chemokine ligand (CCL)19 and CCL21, as well as CXC-chemokine ligand (CXCL)12 and CXCL13 41. These lymphoid chemokines orchestrate the lymphocyte homing and compartmentalisation of the lymphoid organs. CCL19 and CCL21 are produced by stromal cells and regulate the homing of CCR7+ naive T-cells and mature DCs to T-cell areas. CXCL12 and CXCL13 are produced by FDC, thought to originate from mesenchymal stromal cells, and attract CXCR5+ B-cells into the follicles. Activated tissue fibroblasts (e.g. fibroblast-like synoviocytes) share several features with the stromal cells that form the reticular network of secondary lymphoid organs, including the production of lymphoid chemokines (such as CXCL12, formerly called stromal cell-derived factor 1).

{kind=link}
{kind=link}
{kind=link}
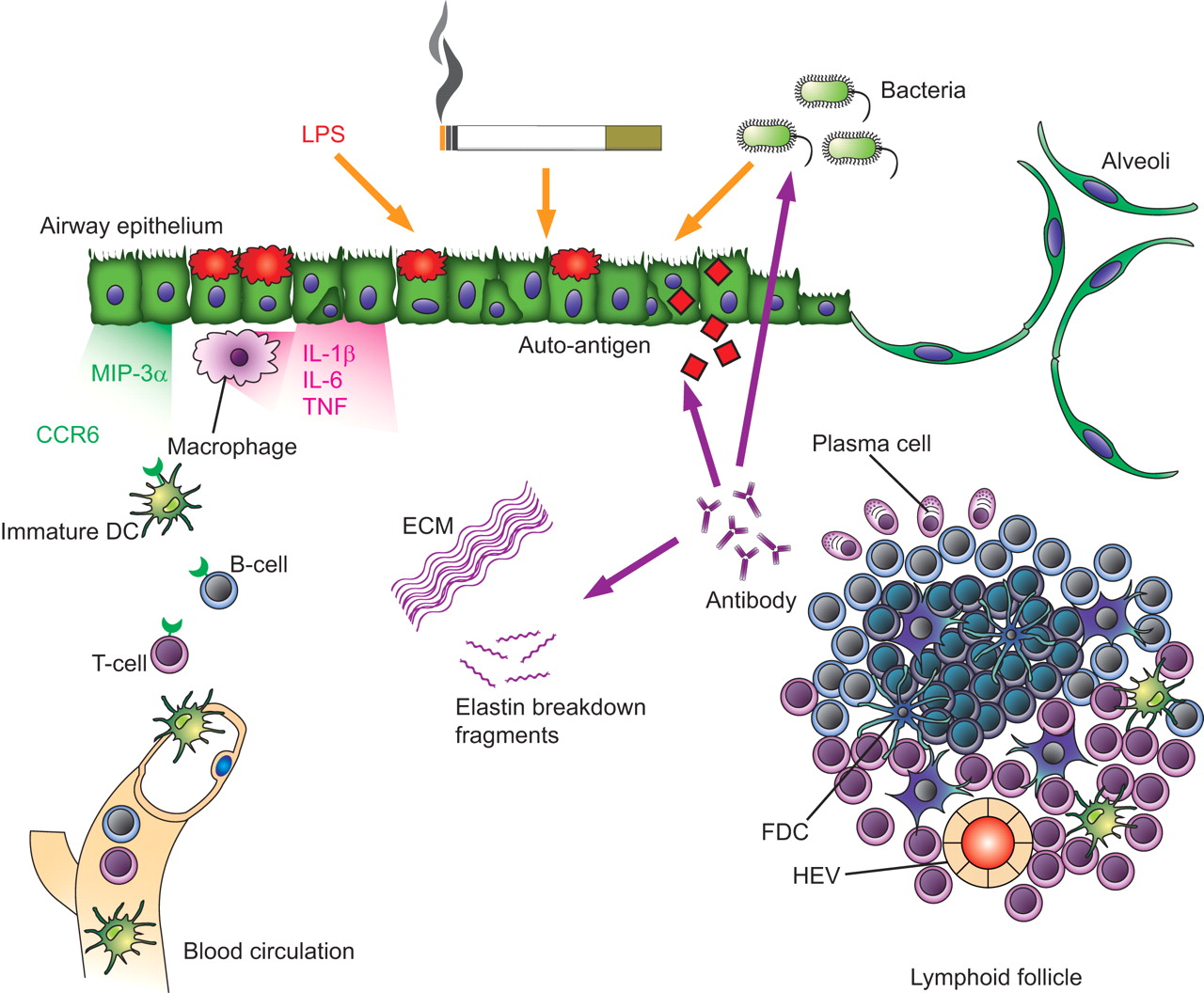
Development of lymphoid follicles in chronic obstructive pulmonary disease. In response to environmental insults, such as lipopolysaccharide (LPS), cigarette smoke and bacteria, airway epithelium and macrophages express cytokines (including interleukin (IL)-1β, IL-6 and tumour necrosis factor (TNF)-α) and chemokines (e.g. CCL20/macrophage inflammatory protein (MIP)-3α), leading to the accumulation of immature dendritic cells (DCs), T-cells and B-cells. When inflammation becomes chronic due to persisting antigen exposure and/or tissue damage, lymphocyte aggregates will give rise to organised lymphoid follicles with separated B- and T-cell areas. Mature lymphoid follicles may contain high endothelial venules (HEV), follicular dendritic cells (FDC) and germinal centres with the potential to produce plasma cells and antibody responses against bacteria and/or auto-antigens, such as breakdown fragments from the extracellular matrix (ECM). Inhaled and systemic corticosteroids may interfere with several mechanisms involved in the development and function of lymphoid follicles.
DCs
In the respiratory tract, myeloid DCs form a dense network of antigen-presenting cells in or just beneath the epithelium and survey the airways for exogenous and endogenous danger signals 42. Recently, a significant increase in Langerin+ DCs has been demonstrated in the small airways of patients with COPD compared with healthy smokers and never-smokers 43. As in the gastrointestinal tract, the accumulation of immature Langerin+ DCs was associated with increased CCL20 levels in induced sputum and lung tissue of COPD patients 43. Upon antigen uptake and activation, DCs lose all chemokine receptors except CCR7, which regulates the migration of DCs towards the regional lymph nodes as well as the influx of naïve T-cells into the paracortex. It is conceivable that similar mechanisms also direct the migration of DCs and T-cells towards the T-cell zone of lymphoid follicles (fig. 3⇑).
PATHOPHYSIOLOGICAL ROLE OF LYMPHOID FOLLICLES IN COPD
What are the antigenic stimuli that trigger the formation of lymphoid follicles?
There are several mechanisms that may underlie the increased numbers of B-cell follicles in lung tissue of COPD patients. The B-cell follicles could reflect, in part, a nonspecific, polyclonal, proliferation of B-cells, induced by local production of B-cell activating mediators including cytokines (IL-4, IL-6, IL-13, TNF-α and lymphotoxin) . Alternatively, supported by the demonstration of (oligo)clonality in follicular B-cells in the lung, they might well be the result of a specific humoral immune reaction 11. At present it is unclear against which antigen(s) this B-cell proliferation could be directed. At least three potential sources of antigens should be considered: 1) microbial; 2) CS components or derivatives; and 3) auto-antigens, encompassing epithelial (neo)antigens and degradation products of extracellular matrix.
Chronic bacterial and viral colonisation and/or infection
The increase in lymphocytes in the small airways and their organisation into bronchial lymphoid follicles are consistent with an adaptive immune response, which is believed to develop in relation to chronic bacterial and/or viral colonisation and infection of the lower airways 44. Several observations plead in favour of this hypothesis.
First, colonisation of the lower airways by bacteria is a common feature in COPD and, therefore, it is possible that at least part of the reactivity of B- and T-cells in the lymphoid follicles is directed against bacterial antigens 44. Moreover, microbial colonisation and infection occur more frequently in the later stages of COPD (severe and very severe COPD) than in milder disease; this parallels the increasing frequency of peribronchiolar lymphoid follicles in more severe COPD. Up to 52% of patients with severe COPD are colonised in the stable state with possible bacterial pathogens, including Haemophilus influenzae, Streptococcus pneumoniae, Moraxella catarrhalis and Pseudomonas aeruginosa 45.
In addition, patients with COPD are prone to frequent exacerbations which are a significant cause of morbidity and mortality. The frequency of COPD exacerbations is modulated by the degree of airway bacterial colonisation in the stable state, and by acquisition of new strains within a pathogenic bacterial species 44, 46, 47. Using an in vivo model of airway infection in mice, strains of H. influenzae isolated from COPD patients during exacerbation induced more airway inflammation than colonisers 48. Importantly, exacerbation strains are strains which are not only isolated for the first time from sputum of patients experiencing clinical exacerbation symptoms, but are also accompanied by the appearance of new bactericidal antibodies to the homologous infecting strain (compared with serum obtained 1 month before exacerbation). In a cohort study of COPD patients investigating the dynamics of carriage and immune responses to M. catarrhalis, Murphy et al. 49 demonstrated that asymptomatic colonisation was associated with a greater frequency of a sputum IgA response than exacerbation, whereas the intensity of the serum IgG response was greater after exacerbations than colonisation. Since development of new sputum IgA and serum IgG were not correlated, mucosal and systemic immune responses appear to occur independently. Moreover, M. catarrhalis was cleared efficiently from the respiratory tract after a short duration of carriage, indicating that COPD patients develop strain-specific protection. In contrast, antibody responses to P. aeruginosa developed in 54% of COPD patients with persistent carriage, suggesting that antibody responses to this bacterium do not mediate clearance, but are a marker for colonisation 50. Thus, the functional role of the B-cell response to airway bacteria in COPD appears to be strain-dependent.
Secondly, in vivo studies in mice have demonstrated that peribronchial lymphoid aggregates and/or follicles can be induced by live infection with influenza virus or repeated exposure to bacterial lysates of NTHi or bacterial cell wall components (LPS) 31–33. These animal studies indicate that the chronic exposure of the airways to bacteria and/or bacterial products may be sufficient to induce lymphoid neogenesis. However, van der Strate et al. 11 elegantly demonstrated that this is not a prerequisite, since the mice that were chronically exposed to CS and developed B-cell follicles were kept under specified pathogen free conditions, together with the air-exposed littermates that did not have these lymphoid follicles. Extensive analysis of fresh frozen samples of the lungs of these mice by RT-PCR did not reveal the presence of microbial species (including Mycoplasma, Chlamydia, adenovirus or Pneumocystis).
Degradation products of extracellular matrix
Apart from microbial colonisation and infections as the trigger of the development of lymphoid follicles in the lung, an antigen specific immune response against neo- or self antigens present in lung tissue could also be the trigger. In the lungs of COPD patients there is a chronic inflammatory response present with the ongoing recruitment of inflammatory cells and concomitant lung tissue damage. This inflammatory environment can provide the optimal conditions for B-cells to form aggregates and start or sustain a local immune response. This immune response can be directed against lung matrix proteins of the destructed tissue, which can be recognised as neo-antigens. It is known that degradation products of extracellular matrix, such as hyaluronic acid, collagen and elastin, have a chemotactic effect on neutrophils and monocytes 51, 52. The effects mediated by the inflammatory cells may lead to ongoing degradation of extracellular matrix, which results in maintenance of the chemotactic gradient and therefore inflammation. It is conceivable that B-cells are either locally present or attracted by this gradient, and start producing antibodies against the extracellular matrix degradation products. This is supported by the recent findings of Lee et al. 53, showing the presence of anti-elastin antibodies in plasma and anti-elastin antibody producing B-cells in lung tissue of emphysema patients. The produced antibodies subsequently bind to the fragments of the extracellular matrix, but also to the intact extracellular matrix and cause degradation of extracellular matrix by “frustrated phagocytosis”. The follicles in COPD lung tissue resemble the development of similar B-cell follicles in the inflamed synovia in rheumatoid arthritis 54.
The immune response could also be directed against CS components or derivatives which are precipitated in lung tissue, since a low number of these follicles can be observed in “healthy smokers” 13. CS contains ∼4,500 different compounds 55, of which it is likely that several are immunogenic (e.g. tobacco glycoprotein). These compounds will precipitate in the lung, possibly partially bind to the extracellular matrix and may elicit an antibody response. Subsequently, these newly produced antibodies will bind to their antigens, forming an immune complex. Phagocytic immune cells generally remove antigen–antibody complexes. If these complexes are either bound to the extracellular matrix, or in the vicinity of it, the extracellular matrix may subsequently be broken down as an “innocent bystander” due to the inflammatory reaction against the immune complexes by these phagocytic cells, similar to rheumatoid arthritis.
Both the mechanisms described for CS- or extracellular matrix-derived antigens may also explain the observation that pulmonary inflammation continues in COPD patients after smoking cessation 2–4. Even though the primary stimulus for inflammation (CS) has vanished, the inflammation continues: it is well known that at least some tobacco residues remain present in the lung for a long time. Direct or indirect antibody-mediated degradation of extracellular matrix results in newly formed extracellular matrix-fragments, which may elicit further inflammation.
Hypothesis linking microbial and auto-antigen pathways
Notwithstanding the notion that the original response of these B-cell follicles in COPD might be protective against bacteria colonising or infecting the lower airways, continued presence of these local immune responses in the lung and the concomitant tissue destruction can promote the breaking of immune tolerance by, for instance, epitope spreading, molecular mimicry or bystander activation (see Appendix). These processes are described to occur after persistent microbial infection or chronic inflammation with the presence of tissue damage. The immune response then extends from the original inducing antigen to similar, often self-reactive epitopes, leading to increased and perpetuated destruction of the inflamed organ and to the initiation of autoimmune responses. In several autoimmune diseases, where similar lymphoid follicles have been shown to be present, these processes have been implicated to play a role in the initiation of the autoimmune response based on findings in animal models. However, there is no direct evidence yet for such an autoimmune mechanism in COPD. Nevertheless, the presence of the chronic inflammatory response in the lungs of COPD patients together with the occurrence of microbial colonisation and recurrent infections, and the presence of lung tissue destruction might provide the circumstances for these processes to occur.
Are lymphoid follicles in COPD protective or harmful?
Depending on the inducing factors of these lymphoid follicles in the lung, they can be regarded as protective, e.g. protection against infections, or harmful, e.g. by producing auto- antibodies and contributing to the perpetuation of the ongoing inflammatory response.
Protection against bacterial and viral colonisation and/or infection
Lymphoid neogenesis might have a protective function by inducing a local (and systemic) immune response against the infectious agents. Indirect evidence in favour of a protective role of B-cell follicles in COPD comes from a randomised controlled trial investigating the efficacy and safety of rituximab in patients with COPD. Rituximab is a chimeric human/mouse monoclonal antibody directed against the CD20 antigen, expressed on pre-B-cells and mature B-cells, but not on other cells (including plasma cells). It induces lysis and apoptosis of normal and malignant human B-cells. Rituximab has been long approved for treatment of CD20+ B-cell non-Hodgkin’s lymphoma 56, 57, and has recently been approved for treatment of rheumatoid arthritis in patients who have failed TNF-inhibitor therapy 58, 59. The study with rituximab in COPD has been terminated early by the data safety monitoring board because of a significant increased risk of infectious complications (including lower respiratory tract infections) in COPD patients treated with rituximab compared with placebo.
Harmful?
Recently, an increased presence of circulating memory B-cells in peripheral blood of current smokers has been demonstrated, suggesting a smoke-induced specific immune response. Moreover, given the possibility of epitope spreading, molecular mimicry or bystander activation (see Appendix) in COPD, the humoral immune response is likely to be polyreactive and can thus be directed against several epitopes among which are other cellular structures of the inflamed lung. Recently, a high prevalence of anti-Hep-2 epithelial cell auto-antibodies in COPD patients was shown, together with a higher presence of antibodies against pulmonary airway epithelial cells in COPD patients compared to healthy controls 60, 61. Additionally, Taraseviciene-Stewart et al. 62 developed a model of autoimmune emphysema in rats by injection of xenogeneic endothelial cells, and recently showed the presence of anti-endothelial cell antibodies in patients with COPD 63, 64. Whether these antibody responses in COPD are pathogenetic and/or true autoimmune responses is still to be determined.
An important characteristic of a pathogenetic autoantibody would be the induction of disease after passive transfer. In the rat model of autoimmune emphysema this was proven by adoptive transfer of the anti-endothelial cell antibodies 62. For the anti-elastin and anti-epithelial antibodies detected in COPD patients, however, this has not yet been proven and should be addressed in similar animal models. Another characteristic of a pathogenetic autoantibody is the presence in or near the disease specific lesion. This characteristic has not been directly proven in COPD. However, Lee et al. 53 showed the presence of anti-elastin producing B-cells in lung tissue, implying that these antibodies are produced locally in the lung. Additionally, Feghali-Bostwick et al. 60 showed the deposition of IgG immune complexes and complement (C3) in the lungs of COPD patients and demonstrated in vitro that antibodies from COPD patients can exert antibody-mediated cellular cytotoxicity. Both studies are in support of a pathogenetic role for these antibodies in COPD.
INFLUENCE OF PHARMACOTHERAPY ON LYMPHOID FOLLICLES
Clinical data on the effect of pharmacotherapy on lymphoid follicles are very scarce and indirect, due to the requirement of invasive procedures to obtain relatively large lung samples for pathological examination. In the NETT, patients with severe (GOLD 3) and very severe (GOLD 4) COPD were studied longitudinally after lung volume reduction surgery 65. When the pathological changes of the small airways in the resected lung specimens were (cor)related with clinical outcomes after surgery, the investigators found that occlusion of the small airways by inflammatory exudates containing mucus was associated with early death in these patients with severe emphysema 66. Intriguingly, there was a strong trend toward a reduction in the number of airways containing lymphoid follicles in COPD patients receiving oral and/or inhaled corticosteroids. The observation that steroid therapy is associated with a reduction in the numbers of follicle-containing airways suggests that steroid-induced suppression of the adaptive immune responses in the peripheral airways might increase the probability of infection in the lower respiratory tract, and may account for the increase in pneumonias observed in recent clinical trials, including TORCH 67 and INSPIRE 68, as well as in observational studies 69. However, these observations should be interpreted with caution due to the cross-sectional nature of the pathological evaluation in NETT.
We put forward the hypothesis that the increased risk of pneumonias in corticosteroid-treated patients with COPD is partially due to the steroid-induced suppression of adaptive immune responses in general, and of pulmonary B-cell lymphoid follicle formation and function in particular. Several mechanisms might contribute to the suppression of lymphoid follicles by corticosteroids. First, inhaled corticosteroids have profound effects on the airway epithelium, which normally facilitates adaptive immune responses by the synthesis of a wide array of lipid mediators, cytokines and chemokines. Central among the anti-inflammatory properties of corticosteroids is the ability to blunt the infiltration of inflammatory cells, including B- and T-cells, within the airways and lungs through the potent suppression of epithelial-derived cytokines and chemoattractants 70 (fig. 3⇑). Importantly, recent insights into the mechanisms of corticosteroid action in airway epithelium have highlighted a concomitant protective action of corticosteroids on innate immune responses 71, 72 Secondly, corticosteroids also inhibit inflammatory cell migration to sites of inflammation by inhibiting the expression of adhesion molecules on leukocytes and endothelial cells. Thirdly, corticosteroids inhibit epithelial-derived signals (e.g. granulocyte-macrophage colony-stimulating factor and thymic stromal lymphopoietin), which normally enable recruitment, survival and activation of DCs. Upon activation, DCs mature and migrate to regional lymph nodes and presumably also lymphoid follicles, inducing T-cell activation and differentiation (see above) which is modulated by treatment with corticosteroids 73. Finally, corticosteroids also inhibit T- and B-cell responses directly, by multiple complex mechanisms, including induction of apoptosis in lymphocytes, modulation of B-cell differentiation and inhibition of the expression of effector cytokines (in particular the T-helper 2 cytokines IL-4, IL-5 and IL-13) in T-cells. All these mechanisms may contribute to the tentatively suppressive effects of corticosteroids on the development and function of lymphoid follicles in COPD patients treated with inhaled or systemic corticosteroids (fig. 3⇑).
CONCLUSION AND FUTURE PROSPECTS
Pathogenesis and progression of severe-to-very severe COPD, in particular, is associated with the presence of peribronchial and parenchymal lymphoid follicles. Using in vivo models of COPD, lymphoid follicles have been described in the lungs of mice which were either chronically exposed to CS or repeatedly exposed to Gram-negative bacteria or LPS. In humans, pulmonary lymphoid neogenesis is not only observed in patients with COPD but also in other chronic diseases of the airways and lungs, including bronchiectasis, follicular bronchiolitis, LIP and pulmonary fibrosis. Outside the lung, lymphoid follicles have been demonstrated in several infectious and autoimmune diseases. Moreover, in some autoimmune diseases, the responsible auto-antigen has been detected within these ectopic lymphoid tissues. The cellular and molecular mechanisms involved in the development of lymphoid follicles are similar to those responsible for the organisation of lymph nodes during organogenesis. The antigenic stimuli that trigger the formation of B-cell follicles in COPD may include microbial antigens, CS components or derivatives, and auto-antigens, encompassing degradation products of extracellular matrix and epithelial (neo)antigens. Epitope spreading, molecular mimicry and bystander activation of B-cells might link the antimicrobial and autoimmune antibody responses observed in patients with severe COPD and emphysema. Therefore, the lymphoid follicles and their ensuing local immune responses might be regarded as either protective (e.g. protection against colonisation and/or infection of the lower respiratory tract) or harmful (e.g. by producing auto-antibodies and perpetuating the ongoing pulmonary inflammation).
We are convinced that the study of the development, structure and function of lymphoid follicles in COPD will provide important clues to a better understanding of the pathogenesis of COPD, including the aetiological agents involved and the mechanisms responsible for the persistent airway inflammation and the relentless progression of the disease. Important questions regarding the role of B-cell follicles and the ensuing antibody responses remain to be addressed. First, since COPD is a heterogeneous disease, encompassing chronic bronchitis, small airways disease (obstructive bronchiolitis) and pulmonary emphysema, does the contribution of lymphoid follicles to the pathogenesis differ between the different compartments they are found in? More research is needed to study the relative contribution of lymphoid follicles in the pathogenesis of the different (e.g. clinical, radiological, physiological and pathological) phenotypes of COPD 74–76. Secondly, does the presence of auto-antibodies implicate that COPD is really an autoimmune disease, where B-cell responses drive the pathogenetic mechanisms, or are these auto-antibodies only an epiphenomenon occurring late in the course of the disease (i.e. in end-stage COPD) and, thus, should they be regarded as innocent bystanders? Are bacterial colonisation and/or infection of the lower respiratory tract a prerequisite for the development of lymphoid follicles in patients with COPD? Is there any link between the systemic manifestations of COPD and the development of lymphoid follicles? What is the effect of current and future pharmacotherapy for COPD on lymphoid follicles? Finally and most importantly, are pulmonary B-cell follicles and responses a worthwhile target in the treatment of (specific subgroups of) COPD patients?
Support statement
This work was supported by the Concerted Research Action of the University of Ghent (Ghent, Belgium; BOF/GOA 01251504) and by the Netherlands Asthma Fund. K.R. Bracke is a postdoctoral fellow sponsored by the Fonds voor Wetenschappelijk Onderzoek–Vlaanderen (Brussels, Belgium).
Statement of interest
A statement of interest for G.G. Brusselle can be found at www.erj.ersjournals.com/misc/statements.dtl
Glossary of terms
- Received October 3, 2008.
- Accepted January 17, 2009.
- © ERS Journals Ltd
References











Jump To
- Article
- Abstract
- LYMPHOID FOLLICLES IN COPD
- LYMPHOID NEOGENESIS IN MURINE MODELS OF COPD
- LYMPHOID NEOGENESIS IN OTHER DISEASES
- CELLULAR AND MOLECULAR MECHANISMS OF LYMPHOID (NEO)GENESIS
- PATHOPHYSIOLOGICAL ROLE OF LYMPHOID FOLLICLES IN COPD
- INFLUENCE OF PHARMACOTHERAPY ON LYMPHOID FOLLICLES
- CONCLUSION AND FUTURE PROSPECTS
- Support statement
- Statement of interest
- References
- Figures & Data
- Info & Metrics