





Abstract
In the lungs, parenchymal and vascular remodelling share pathomechanisms that may explain the relatively high prevalence (30–40%) of pulmonary hypertension (PH) in interstitial lung disease (ILD) patients. Notably, PH significantly contributes to exercise limitation and dismal prognosis of ILD patients. The absence of specific clinical symptoms commonly leads to delayed diagnosis. Besides clinical judgment and out-of-proportion reduction in diffusing capacity, severe hypoxaemia or exercise oxygen desaturation, echocardiography and biomarkers such as B-type natriuretic peptide (BNP) and N-terminal pro-hormone BNP are potentially helpful tools in identifying PH. However, right heart catheterisation is still necessary to confirm the diagnosis. Management of PH in ILD comprises treatment of the underlying disease process and long-term oxygen therapy. Affected patients should be listed for lung transplantation without delay, when appropriate. However, due to age and comorbidities only a minority of ILD patients will be eligible for lung transplantation. In the absence of satisfactory therapies for many ILDs, and considering the clinical burden of PH in affected patients, specific vasomodulatory therapies presently approved for PAH may be promising options for ILD patients. Consequently, there is an urgent need for adequately designed clinical trials to assess the effectiveness of specific PH therapy in the context of ILDs.
- Connective tissue disease
- interstitial lung disease
- pulmonary fibrosis
- pulmonary hypertension
- sarcoidosis
SERIES "PULMONARY HYPERTENSION: BASIC CONCEPTS FOR PRACTICAL MANAGEMENT"
Edited by M.M. Hoeper and A.T. Dinh-Xuan
Number 3 in this Series
Interstitial lung diseases (ILDs) comprise a heterogeneous group of diseases with common functional characteristics (restrictive physiology and impaired gas exchange) and a common final pathway, eventually leading to irreversible fibrosis 1–4. However, inflammation, granuloma formation and fibroproliferation vary considerably between the different entities 2. Development of pulmonary hypertension (PH) in the context of ILDs, conversely, is a well-recognised complication of various ILDs, but has not yet been studied extensively 5–9. New pathogenetic concepts of pulmonary fibrosis share interesting aspects with pathogenetic mechanisms implicated in the development of PH 10–14. In particular, the roles of the endothelin (ET) system, transforming growth factor (TGF)-β1, connective tissue growth factor (CTGF) activation and oxidative stress are well recognised in both conditions, with obvious interrelationships 15–19.
Clinically, PH may impose dyspnoea, fatigue and exercise limitation, symptoms which are also characteristic for ILDs. Consequently, the diagnosis of PH may be missed in ILD patients until signs of right heart failure develop 4, 20–25. Moreover, specific and effective treatment for pulmonary arterial hypertension (PAH) has become available only recently and the previous lack of therapeutic options has also certainly abrogated the necessity of identifying ILD patients with associated PH. During the last decade, however, several specific mechanistic pathways, e.g. prostacyclin I2, ET receptor antagonists and phosphodiesterase inhibitors, have been identified, which allows application of specific pharmacological interventions for the improvement of pulmonary haemodynamics and exercise capacity in patients with PAH 5. Consequently, the role of PH in the natural history of ILDs and the potential benefits of specific treatments of this condition have attracted new attention. Impact of fibroproliferation on the pulmonary vasculature and, conversely, impact of angiogenesis on pulmonary fibrosis, have also been discussed in the context of the pathogenesis of ILDs such as idiopathic pulmonary fibrosis (IPF), implicating an even more fundamental role of vascular biology in the aetiology and pathogenesis of ILDs 26, 27.
The present manuscript summarises the current knowledge about pathogenesis, prevalence, diagnosis, prognosis and management of PH in various ILDs, especially focusing on sarcoidosis, collagen vascular diseases and IPF.
BASIC ASPECTS OF PULMONARY FIBROSIS AND PH
In the area of ILDs, new pathogenetic concepts have evolved recently that primarily relate to IPF but may also have implications for other forms of pulmonary fibrosis, including connective tissue diseases (CTDs) and granulomatous diseases such as sarcoidosis 3, 28–30. According to this new hypothesis, a repetitive injury leads to damage of epithelial cells and basement membranes, followed by exudation of fibrin and focal fibroblast activation and proliferation, finally resulting in fibrotic remodelling of lung parenchyma. Pathophysiological mechanisms that may be involved include the following. 1) Oxidant–antioxidant imbalance, and more specifically a lack of glutathione, which promotes fibrogenesis and inhibits vasodilation 31–42. 2) Formation of alveolar fibrin clots due to impaired intra-alveolar fibrinolysis providing a lead structure for fibroblast chemotactic migration and proliferation as well as for neovascularisation 29, 30, 43. 3) Growth factors such as TGF-β, insulin-like growth factor-1, platelet-derived growth factor (PDGF) and CTGF play a crucial role in the expansion of connective tissue and vascular remodelling in the lungs 44–51. 4) Epithelial cell apoptosis and impaired epithelial regeneration and epithelial-to-mesenchymal transdifferentiation linked to integrin signalling promotes fibrotic tissue remodelling 52, 53. 5) Angiogenesis and neovascularisation; integral components of fibrotic tissue remodelling 26, 27, 53, 54. 6) Mesenchymal stem cells and circulating progenitor cells involved in repair mechanisms after lung injury, which may contribute to the process of lung fibrosis 53–58. 7) Mutations of the telomerase gene, the ATP-binding cassette transporter A3, and surfactant protein-C as causes of familial interstitial lung disease have shed some light on the role of “endoplasmic reticulum stress” and apoptosis as potential aetiological factors of ILDs 59–64.
With respect to the interrelationship of fibroproliferation and PH, animal models are lacking. However, there are some observations that may serve as clues to the missing link. One of these is unbalanced oxidative stress, which has been implicated in the disease process of fibroproliferation and in antivasodilatory and pro-proliferative activities in PH 39–42. Oxidants and a lack of the antioxidant glutathione are linked to apoptosis of alveolar epithelial cells, proliferation of fibroblasts and synthesis of extracellular matrix, and may jointly lead to pulmonary fibrosis. Recent data demonstrate that the ubiquitous soluble guanylyl cyclase (sGC), which is essential for the nitric oxide (NO)–sGC–cyclic guanosine monophosphate (cGMP) signal transduction pathway, is inactivated by oxidative stress and reconstitutes with antioxidants 42. In endothelial cells the NO–sGC–cGMP pathway mediates potent vasodilatory and antiproliferative effects, but profibrotic effects have also been detected in the kidney and liver 42.
Another unifying element in the pathogenesis of pulmonary fibrosis and PH may be the ET system, with ET-1 as the most potent pulmonary vasoconstrictor, which has been detected in elevated concentrations in idiopathic PAH but also in CTDs such as systemic sclerosis (SSc), in IPF with and without associated PH, and in pulmonary sarcoidosis 15, 16, 18, 65–70. In addition to its vasoconstrictor properties, ET-1 is also a potent growth factor for endothelial cells and myofibroblasts 15, 16, 71–73. In animal models, overexpression of ET-1 is associated with enhanced pulmonary parenchymal fibrosis 48, whereas the dual ET receptor A and B antagonist bosentan ameliorates bleomycin-induced pulmonary fibrosis 74. Moreover, recent reports suggest that ET-1 also induces fibrogenesis via interaction with matrix metalloproteinases and initiates epithelial to mesenchymal transition via an ET type A receptor-mediated induction of TGF-β1 75–77. According to this concept of mutual interaction of pathomechanisms related to ILD and PH, circulating factors as well as overspill of locally released mediators may be involved in the pathogenetic process.
Interestingly, the dual ET receptor type A and B antagonist bosentan is already approved for the treatment of various types of PAH and is currently being evaluated for the treatment of IPF. In a feasibility study, bosentan was evaluated by multiple inert gas elimination technique in IPF patients and demonstrated no evidence of increased intrapulmonary shunting 78. A recently published phase II randomised, placebo-controlled trial did not show a positive effect on the primary end-point, 6-min walk distance, in patients with IPF. However, a positive trend was observed with respect to the pre-defined secondary end-point: time to disease progression or death 79. Interestingly, this trend was statistically significant in the subgroup of patients with biopsy-proven IPF 79. These results may indicate that IPF is responsive to bosentan treatment only in the absence of significant honeycombing in the lungs 79. Since clinically significant PH was an exclusion criterion in the study, the beneficial effect may be attributable to antifibrotic activity of bosentan mediated by inhibition of ET.
An illustration of selected pathogenetic mechanisms that may be involved in both pulmonary fibrosis and PH is shown in figure 1⇓.

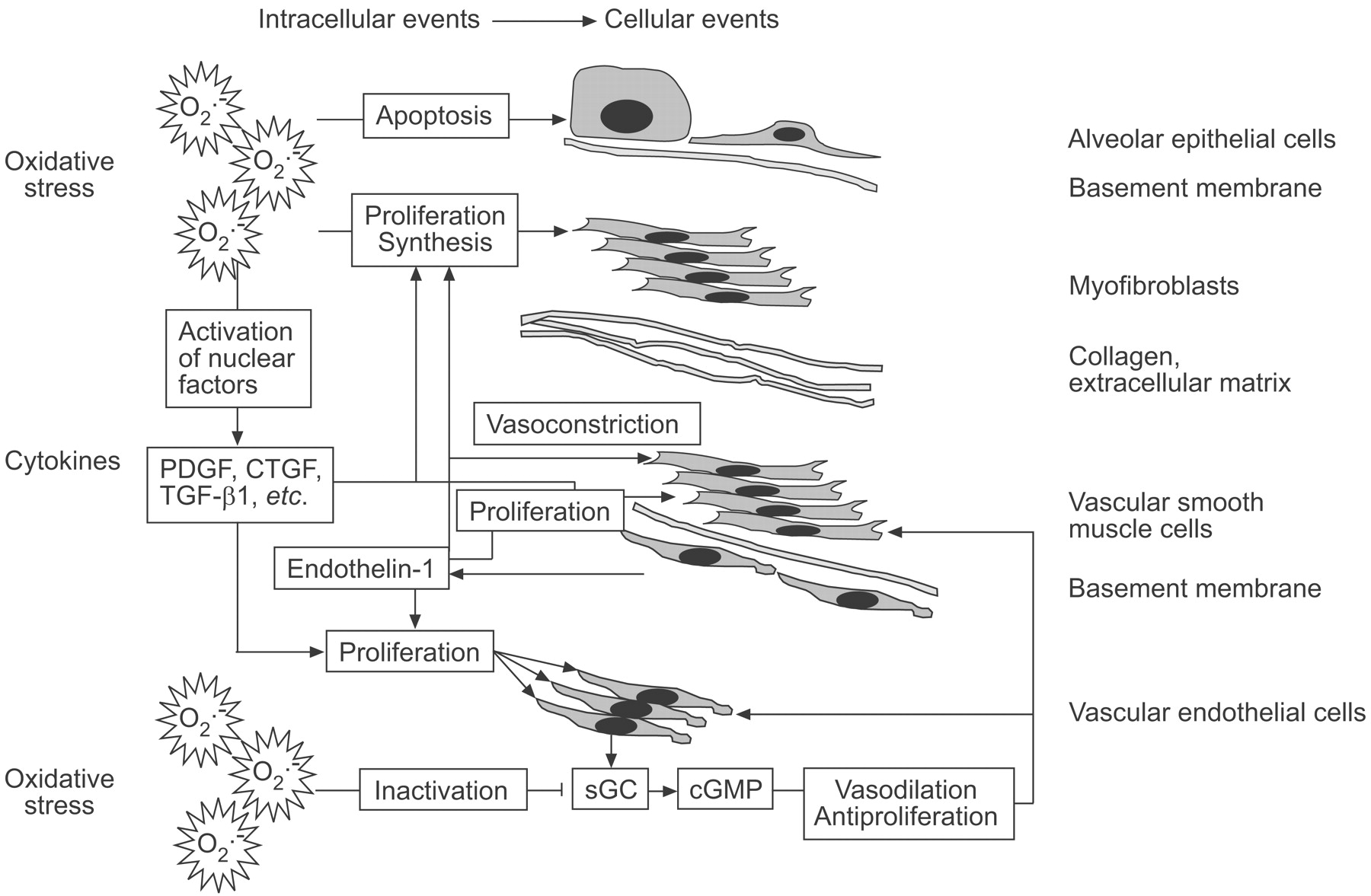{kind=link}
Potential common pathomechanisms of parenchymal and vascular remodelling in interstitial lung disease and pulmonary hypertension. For further explanations see text. PDGF: platelet-derived growth factor; CTGF: connective tissue growth factor; TGF: transforming growth factor; sGC: soluble guanylyl cyclase; cGMP: cyclic guanosine monophosphate.
Besides these “unifying mechanisms” of parenchymal and vascular remodelling, there are also mechanisms that are unique to one or another process. In SSc, for example, gene polymorphisms of the CTGF and stimulatory autoantibodies of the PDGF receptor have been described, which raise the possibility of unique pathogenetic mechanisms, e.g. those driven by autoimmunity in this disease 80, 81. Consequently, immunosuppressive therapy has been shown to be of some, although limited, benefit in SSc-associated ILD 82. However, imatinib, an inhibitor of PDGF-mediated tyrosine kinase signalling, may also offer a promising therapeutic approach to fibrotic lung disease, especially in SSc patients 83, 84.
PREVALENCE AND BURDEN OF PH IN ILD
PH is generally defined as a mean pulmonary artery pressure (Ppa) >25 mmHg at rest or >30 mmHg during exercise. For the purpose of the present review, pulmonary venous hypertension, as indicated by an elevated pulmonary capillary wedge pressure (>15 mmHg), is excluded. The prevalence of PH in various ILDs varies widely according to diagnosis and severity of the lung impairment. The most common ILDs associated with PH, i.e. sarcoidosis, CTDs (especially SSc and rheumatoid arthritis) and IPF will be discussed in the current paper.
Sarcoidosis
In sarcoidosis patients, a recent cross-sectional study based on Doppler echocardiography (using an estimated systolic Ppa ≥40 mmHg as cut-off) reported a prevalence of PH to be 5.7% 85. In contrast, in sarcoidosis patients who were listed for lung transplantation at United Network for Organ Sharing, 73.8 % had PH as assessed by right heart catheterisation (RHC) 86–88. Moreover, PH in sarcoidosis patients listed for transplantation was associated with increased mortality while on the waiting list 87, 88. Interestingly, in studies by Shorr and co-workers 87, 88, neither spirometric testing nor the need for corticosteroids was predictive of PH in sarcoidosis patients, whereas need for supplemental oxygen was an independent predictor of PH on multivariate analysis. Irrespective of other physiological measures, the presence of PH is a predictor of mortality in sarcoidosis patients 85–88. Notably, physiological impairment is not, or at least not closely, related to the development of PH. As PH eventually develops in sarcoid patients in the absence of gross parenchymal abnormalities, parenchymal scarring and hypoxic vasoconstriction cannot be the only mechanisms. Pulmonary artery stenosis due to compression by enlarged lymph nodes and direct granulomatous involvement of the arterial walls are additional pathogenetic factors that have to be taken into account 89–91.
Systemic sclerosis
A recent Japanese study documented isolated restrictive lung physiology suggestive of ILD in 22.5% of SSc patients, isolated PH identified by echocardiography in 19.2%, and a combined restrictive ventilatory deficit along with PH in 18.2% 92. In a cohort of patients with diffuse cutaneous SSc, Trad et al. 93 observed a prevalence of 60% for ILD, 21% for PAH and a combination of ILD along with PH was found in 22.1% 93. PH was also found to be a risk factor for survival, independent of ILD. Again, there was no close correlation between the severity of restrictive ventilatory defect and PH 93.
Idiopathic pulmonary fibrosis
Prevalence of PH, as assessed by echocardiography, is reported in up to 84% of IPF patients. However, these data refer to a pre-selected patient cohort, with echocardiography performed according to the discretion of the investigators 94. In heterogeneous groups of patients with fibrotic lung diseases, with the majority, but not all, suffering from IPF, in a pre-transplant setting, PH is detected by RHC in 28–46% of patients 24, 95. In these studies, again, lung volumes did not correlate closely with Ppa, and the presence of PH was related to decreased diffusion capacity and need for oxygen supplementation. In a recent study, diffusion capacity <40% predicted and mean Ppa >17 mmHg were significant predictors of mortality 95. Moreover, it has been demonstrated that exercise limitation is pronounced in IPF patients with PH, when compared with IPF patients without PH who have equally severe restrictive lung physiology 24, 96.
Conclusion
PH is detected in a significant proportion of patients with various ILDs and, if present, PH is a predictor of mortality. Interestingly, in all three ILDs discussed, restrictive lung physiology was not related to the presence of PH, whereas diffusion impairment (reduced transfer factor for carbon monoxide) and the need for oxygen supplementation were predictors of PH. Moreover, PH appears to pose an additional burden on exercise capacity of ILD patients, independent of abnormalities in lung volumes 96. Consequently, these observations provide a rationale for the specific treatment of PH in the context of ILD as an associated and pathogenetically linked condition, with its own clinical and prognostic sequelae.
DIAGNOSIS AND STAGING OF PH IN ILD
Shortness of breath and exercise limitation are the leading symptoms of both ILD and PH. Consequently, development of PH in a patient with known ILD may be difficult to recognise clinically. Physical signs, such as loud pulmonic component and fixed splitting of the second heart sound, holosystolic murmur of tricuspid regurgitation and diastolic murmur of pulmonic regurgitation, as well as jugular venous distension and right ventricular heave, are generally signs of advanced PH. Early diagnosis of PH in ILD patients before clinical signs of right heart failure develop requires a high index of clinical suspicion. Conversely, even mild PH may significantly impair the prognosis of patients with ILD. In two recent observational studies, mean Ppa >17 and >25 mmHg have been reported to be associated with increased mortality of patients with IPF 24, 97. Therefore, patients with ILD should be screened for coexisting PH.
Radiology
Although standard chest radiography may provide signs of PH, sensitivity of this modality is generally low 98. Enlarged central pulmonary arteries (>15 mm) and elongated retrosternal contact of the right ventricle are typical findings. In computed tomography (CT), enlarged main pulmonary artery (>29 mm) or an increased diameter of the pulmonary artery as compared with the aorta can be documented in patients with moderate to severe PH 99–101. However, early diagnosis of PH is generally not a domain of chest CT. In a recent cross-sectional study of 65 IPF patients with PH (diagnosed by RHC), high-resolution CT failed to demonstrate a correlation between increased diameter of pulmonary arteries (actual measurement or in relation to aortic diameter) and mean Ppa 102.
Echocardiography
In general, the most appropriate method to noninvasively detect PH is transthoracic (Doppler) echocardiography (TTE) 103, 104. Overall, echocardiography was shown to correctly predict PH with sensitivity ranging 0.79–1.0 and specificity ranging 0.6–0.98 6. An estimated systolic right ventricular pressure (or systolic pulmonary artery pressure (SPAP)) of >50 mmHg is accepted to indicate the presence of PH (in the absence of pulmonary valvular stenosis). SPAP values of 35–50 mmHg are considered borderline, whereas SPAP <35 mmHg represents a normal finding 99. However, there are significant limitations of TTE which may lead to over- or underestimation of SPAP: it has been reported that in one-third to one-half of all patients with ILDs valid Doppler echocardiographic measurement of tricuspid regurgitation is not possible for technical reasons 94, 105. Moreover, positive and negative predictive values of TTE were low when compared with RHC results 105. On the contrary, it has been reported that TTE with exposure to hypoxia or stress testing may serve to detect subclinical PH in asymptomatic carriers of gene mutations associated with idiopathic PAH 106.
In summary, it appears that TTE is an important tool for the detection of PH in at least a portion of ILD patients and should, therefore, be employed whenever there is a clinical suspicion of PH. It has to be emphasised that the measurement of the tricuspid regurgitation with Doppler echocardiography alone is not sufficient to address PH in IPF; right ventricular and atrial dimensions as well as eccentricity index and tricuspid annular plane systolic excursion are valuable additional parameters and, of course, a comprehensive assessment of left heart dimensions and function are also imperative. In doubtful cases, RHC remains the gold standard and is strongly advocated.
Natriuretic peptides
Another approach to the diagnosis of PH in ILD is based on the known neurohumoral activation associated with the development of PH that leads to exaggerated release of natriuretic peptides, i.e. atrial natriuretic peptide (ANP), B-type natriuretic peptide (BNP), and N-terminal pro-hormone BNP (NT-pro-BNP) 25, 96, 107–111. In a recent cross-sectional study, it was demonstrated that elevated plasma BNP concentrations are able to detect significant PH (defined as mean Ppa >35 mmHg) in patients with pulmonary fibrosis due to various causes 96. Moreover, BNP and NT-pro-BNP plasma levels were predictive of prognosis in patients with ILD 25, 96. NT-pro-BNP has been found to be more dependent on kidney function, as renal excretion is the only route of clearance for NT-pro-BNP, as opposed to BNP, which also undergoes proteolytic degradation 110. Similarly to BNP, elevated ANP levels were described in patients with PH of various causes, but this biomarker has been studied less frequently 111.
In summary, the available data suggest that BNP and NT-pro-BNP are biomarkers that are helpful for the detection of PH associated with right ventricular dysfunction in patients with underlying ILDs, but these biomarkers probably do not allow for early diagnosis of mild or latent PH. Nevertheless, BNP plasma concentrations have been shown to be significantly correlated to haemodynamic measures, World Health Organization functional class, and 6-min walk distance in patients with ILD and, therefore, may provide valuable information in many ILD patients 96. However, in completely compensated disease, BNP and NT-pro-BNP plasma levels may be normal, despite the presence of significant PH. Moreover, impaired renal function, among other factors, interferes with BNP and NT-pro-BNP plasma concentrations, since both molecules are partially (BNP) or completely (NT-pro-BNP) eliminated by the kidneys 111. Conversely, a normal plasma BNP concentration is associated with a very low probability of PH and with prolonged survival 25, 96. With respect to therapeutic implications, this information may be especially valuable.
Right heart catheterisation
RHC is still the gold standard for the haemodynamic evaluation of the pulmonary circulation and should be performed in every patient with evidence of PH identified by noninvasive investigations, which include clinical findings (systolic murmur suggesting tricuspid insufficiency), out of proportion reduction in diffusing capacity of the lung for carbon dioxide (DL,CO), excessive exercise desaturation and need for high-flow oxygen substitution, radiological signs and TTE findings suggesting PH, as well as elevated BNP or NT-pro-BNP levels 5–7, 99, 112. RHC is necessary to document the severity of PH and potential right ventricular dysfunction. It is also necessary to exclude PH caused by diastolic dysfunction of the left ventricle, which may be difficult to diagnose with echocardiography. In PAH patients, pulmonary vasoreactivity testing is recommended by using inhaled NO or alternative agents, such as epoprostenol or i.v. iloprost, aerosolised iloprost or i.v. adenosine 5–7. For PAH a positive vasodilator test is defined as a decrease in mean Ppa by ≥10 mmHg to an absolute value of ≤40 mmHg with an increased or unchanged cardiac output. Adequate oxygenation during the test has to be maintained. A positive vasodilator test in idiopathic PAH is generally considered to indicate that the respective patient is responsive to high-dose calcium channel blocker (CCB) therapy. This has, however, not been demonstrated for patients with underlying ILDs. Consequently, vasoreactivity testing and high-dose CCB therapy currently has no role in the management of PH in association with IPF and other ILDs.
Pulmonary function test
In general, pulmonary function tests (PFTs) do not specifically contribute to the assessment of PH. However, patients with emphysema in conjunction with IPF may manifest nearly normal lung volumes associated with a severely reduced diffusing capacity as measured by carbon monoxide transfer factor via the single breath method (DL,CO). This discrepancy between relatively well-preserved lung volumes accompanied by very low DL,CO (<35% pred) is associated with a higher probability for the presence of PH 113–115. Recent studies suggest that DL,CO <40% pred and need for oxygen supplementation are predictive of PH in patients with IPF and in sarcoidosis 24, 86–88, 95, 97, 112, 116. Zisman et al. 117 have developed a multivariable linear regression model using arterial oxygen saturation measured by pulse oximetry (Sp,O2) and the ratio of forced vital capacity (FVC) % pred to DL,CO % pred for the prediction of mean PH with a positive predictive value of 71% and a negative predictive value of 81%. Further validation of this approach is needed, in order to confirm its usefulness. In SSc, Steen et al. 118 followed a similar approach and found that DL,CO <55% pred and a ratio of FVC % pred/DL,CO % pred >1.4 were associated with PH; however, only 22% of patients fulfilling this criteria developed PH, in contrast to only 2% without these criteria who developed PH.
In conclusion, low DL,CO and need for oxygen supplementation are associated with a high probability of PH, whereas FVC is less predictive. In particular, disproportionate reduction in DL,CO compared with FVC should raise suspicion of PH, although a firm relationship between FVC and DL,CO that would indicate PH is not yet established.
Histology
Histology is not required to make a diagnosis of PH. Moreover, transbronchial biopsy obtained via bronchoscopy is relatively contraindicated in patients with PH (mean Ppa >30 mmHg), due to increased risk of bleeding 119. Similarly, the morbidity and mortality risk of surgical lung biopsy (video-assisted thoracic surgical or open) is increased in patients with ILD and PH 6. Obtaining a lung biopsy for the sole purpose of assessment of PH in ILD is therefore generally discouraged.
General diagnostic approach
In patients with ILD and suspected PH, the clinician should be aware of the fact that PH in this setting may be due to another cause (e.g. pulmonary embolism or undiagnosed CTD). Therefore, noninvasive investigations should include transthoracic Doppler echocardiography, PFTs including carbon monoxide transfer factor, measurement of BNP or NT-pro-BNP, immunological markers as appropriate, overnight oximetry and CT chest angiography. If these results support the presence of PH, RHC including vasoreactivity testing should be performed.
TREATMENT OF PH IN THE CONTEXT OF ILD
The primary treatment approach is directed to the underlying ILD and therefore requires an accurate diagnosis, which may require surgical lung biopsy for histological differentiation. In most ILDs, immunosuppressive or anti-inflammatory regimes are employed that consist of prednisone eventually combined with a cytotoxic agent, such as azathioprine or cyclophosphamide 120–123. In IPF, preservation of lung function may be enhanced if prednisone and azathioprine is combined with high-dose N-acetylcysteine therapy, which appears to exert antioxidative effects via the replenishment of pulmonary glutathione stores 35, 36, 124. Unfortunately, there are very limited and contradictory reports regarding the effects of immunosuppressive therapy on PH 125–127. Nevertheless, in some reports, positive effects on pulmonary haemodynamics have been described in patients with underlying CTD or sarcoidosis, although these effects seem to be unpredictable and inconsistent.
General treatment recommendations for patients with PH have only been established for PAH patients, which refers to Group I patients of the Venice classification 5–7. These guidelines comprise administration of diuretics in patients with volume overload due to right heart failure, as well as considering digitalis. Oxygenation status should be assessed in these patients at rest and during exercise. Long-term oxygen therapy should be initiated to maintain Sp,O2 ≥90% (≥60 mmHg) at rest and during exercise. Anticoagulation is also recommended and may be beneficial in avoiding pulmonary embolism and in situ thrombosis in these patients, although it has to be noted that adequately designed prospective studies demonstrating a beneficial effect of anticoagulation in PAH are lacking. For PH associated with ILDs there are no treatment guidelines available yet. Nevertheless, as there are clinical, pathogenetic, and pathophysiological similarities between PH associated with ILDs and PAH, similar treatment modalities may be effective. However, this remains to be established, as studies in support of this assumption are not currently available. Only recently, a randomised controlled clinical trial addressing the potential effect of anticoagulation on the survival of IPF patients has been published 128. This latter study was performed in Japan, in IPF patients who were hospitalised due to clinical worsening. Standard therapy with high-dose prednisone along with anticoagulation or standard therapy alone was initiated in a randomised fashion. The primary end-point and major finding was a significant survival benefit for the anticoagulation group 128. However, the design of the trial had significant limitations and did not distinguish whether the observed effect had any association with the presence of PH and/or pulmonary embolism 128. In view of the limited evidence available, the potential benefit and existing drawbacks, including bleeding risks, should be openly discussed with the patients. In well-informed patients who are willing, anticoagulation can be justified, especially in the presence of severe PH with right heart dysfunction associated with ILD.
In ILD patients with severe right heart failure requiring management in the intensive care unit setting, PH may dominate the clinical picture and determine the fate of the patient. In this situation it may be appropriate, after considering the clinical context (including the age of the patient and comorbidities), to treat these patients with vasomodulatory agents. Intravenously administered epoprostenol or iloprost may increase intrapulmonary shunt in ILD patients, thus aggravating hypoxaemia. Therefore, the use of inhaled iloprost or NO, or orally administered agents such as sildenafil or bosentan, may be considered in these acute and life-threatening situations, although these agents have not been approved for this clinical indication 129–133. In addition, volume management, oxygen supplementation, and noninvasive ventilation are applied as needed.
In chronic disease states of patients with ILD and associated PH, the value of specific agents to treat PH is not yet established. In particular, there is no approved therapy for PH in the context of ILD. Considering the pathogenetic links between pulmonary fibrosis and PH on the one hand, and the impact of PH on exercise tolerance and prognosis of patients with PH on the other, it appears plausible that available agents to treat PH could also have a potential to positively affect the outcome of patients with ILD and PH. This is even more tantalising for patients with IPF, for whom no satisfactory therapy is yet available to address the underlying fibrotic process. Interestingly, there are reports of acute effects of inhaled iloprost or sildenafil on pulmonary haemodynamics in patients with pulmonary fibrosis of various aetiologies, which yielded similar results as those described in PAH patients 129, 132. Moreover, initial uncontrolled observations suggest that sildenafil, an orally administered phosphodiesterase-5 inhibitor, improves the 6-min walk distance in IPF patients with PH by a mean of 47 m after 3 months of therapy, which is similar to treatment effects observed in clinical trials of PAH 134.
Given the substantial evidence linking ET to the pathogenesis of both PAH and pulmonary fibrosis, there is a potential for ET antagonists to ameliorate PH in ILD. Consequently, the dual ET receptor A and B antagonist, bosentan, was employed in a recent placebo-controlled clinical phase II trial investigating its effects on IPF in the absence of PH. Although the primary end-point (6-min walk distance) was negative, there were positive signals from secondary end-points and post hoc subgroup analyses that deserve further investigation 79. Indeed, there are also reports suggesting that bosentan may be beneficial in PH associated with sarcoidosis and other ILDs 135–137.
Taken together, there is an unmet clinical need to treat patients with PH in association with ILDs. The available data strongly suggest that PH in ILD is of clinical and prognostic significance and similarities exist in clinical behaviour to that of PAH. However, there is a lack of high-quality clinical trials investigating the treatment of PH in ILD patients with the available pharmacological agents, i.e. prostanoids (e.g. iloprost aerosol), phosphodiesterase inhibitors (e.g. sildenafil) and ET receptor antagonists (e.g. bosentan, ambrisentan or sitaxentan).
LUNG TRANSPLANTATION
According to the current guidelines, PH in ILD has the worst prognosis and is an indication for immediate listing for lung transplantation if the patient is otherwise appropriate for this procedure 138. Accordingly, evaluation for lung transplantation should be considered at first diagnosis in these patients and with special emphasis if the disease is progressive despite medical therapy 138. There is conflicting evidence regarding the type of transplantation which should be performed, i.e. single or double lung transplantation 139–145. The new International Society for Heart and Lung Transplantation report asserts that in IPF long-term outcomes are better for double lung transplantation 145, which is, however, contrary to a previous evaluation of the registry data by Meyer et al. 144. In older patients with pulmonary fibrosis and mild-to-moderate PH, single lung transplantation seems to be particularly appropriate 140, 144. A decision in favour of bilateral lung transplantation in IPF patients has to take into account multiple factors, including severity of PH, infections or colonisations (e.g. aspergilloma in sarcoidosis or IPF), potential complications in the native lung, age of the recipient and experience of the transplant centre. Ultimately, the decision in favour of single or double lung transplantation is always individualised. Finally, living lobar lung transplantation may also be an option in selected patients 145.
FUTURE DIRECTIONS
As has been already described, there are shared mechanisms in the pathogenesis of pulmonary fibrosis and pulmonary hypertension that may be addressed by new therapeutic agents, such as the platelet-derived growth factor inhibitor imatinib or inhibitors of transforming growth factor-β 146. Respective clinical trials are under way. However, further research is clearly needed to decipher the pathways by which pulmonary hypertension arises in patients with interstitial lung diseases. There are likely to be some mechanisms that are peculiar to specific interstitial lung diseases and need to be better understood in order to optimise management of these patients. Emerging understanding of these crucial issues will likely open new therapeutic avenues in treating pulmonary hypertension and the underlying interstitial lung disease.
Statement of interest
A statement of interest for J. Behr can be found at www.erj.ersjournals.com/misc/statements.shtml
Footnotes
-
Previous articles in this series: No. 1: Dupuis J, Hoeper MM. Endothelin receptor antagonists in pulmonary arterial hypertension. Eur Respir J 2008; 31: 407–415. No. 2: Gomberg-Maitland M, Olschewski H. Prostacyclin therapies for the treatment of pulmonary arterial hypertension. Eur Respir J 2008; 31: 891–901.
- Received December 19, 2007.
- Accepted February 24, 2008.
- © ERS Journals Ltd
References










