





Abstract
Histone acetylation and deacetylation promote and repress gene transcription, respectively. Recruitment of histone deacetylases (HDAC) to sites of inflammatory gene transcription has been proposed to explain part of the anti-inflammatory activity of steroids. To examine whether this concept extends to other inflammatory conditions, the current authors investigated the role of histone acetylation and the effects of steroids on the ventilation-induced induction of pro-inflammatory genes.
Isolated perfused mouse lungs were ventilated for 180 min with low peak inspiratory pressure of 10 cmH2O or high peak inspiratory pressure of 22.5 cmH2O (overventilation) and treated with the HDAC inhibitor trichostatin A (TSA), the steroid dexamethasone or both.
Overventilation increased histone acetylation at H4K12, as well as gene and protein expression of tumour necrosis factor (TNF), macrophage inflammatory protein (MIP)-2α and interleukin (IL)-6; these effects were reversed by dexamethasone. In the presence or absence of dexamethasone, TSA enhanced overventilation-induced induction of TNF and MIP-2α, but decreased that of IL-6, indicating that the effects of HDAC are gene dependent.
Thus, H4K12 acetylation and its regulation by steroids may be relevant for inflammatory gene transcription during overventilation. Histone deacetylases appear to play an important gene-dependent regulatory role in this process, with the caveat that histones are not the only substrates of histone deacetylase isoenzymes.
Protein acetylation plays a pivotal role in the regulation of gene transcription. This was first demonstrated in histones that become acetylated at specific lysine residues prior to gene transcription 1. Histone acetylation disrupts higher-order chromatin structure and facilitates the interaction of transcription factors and the transcription machinery with DNA. Histone acetylation is carried out by histone acetyltransferases (HAT) and reversed by histone deacetylases (HDAC), turning gene transcription on and off. Recently, histone acetylation and deacetylation have been proposed as important regulators of inflammatory gene transcription in the lung, in conditions such as asthma and chronic obstructive pulmonary disease (COPD) 1. In particular, it was proposed that inactivation of HDAC by reactive oxygen species (present in cigarette smoke) would inhibit one major anti-inflammatory mechanism by which steroids act, thus explaining the steroid resistance of COPD patients. Ventilator-induced lung injury is another clinically relevant inflammatory response with oxygen radical production in the lung 2–4. By analogy to COPD, it seems possible that modification of histone acetylation modulates the effects of steroids in the setting of ventilator-induced lung injury.
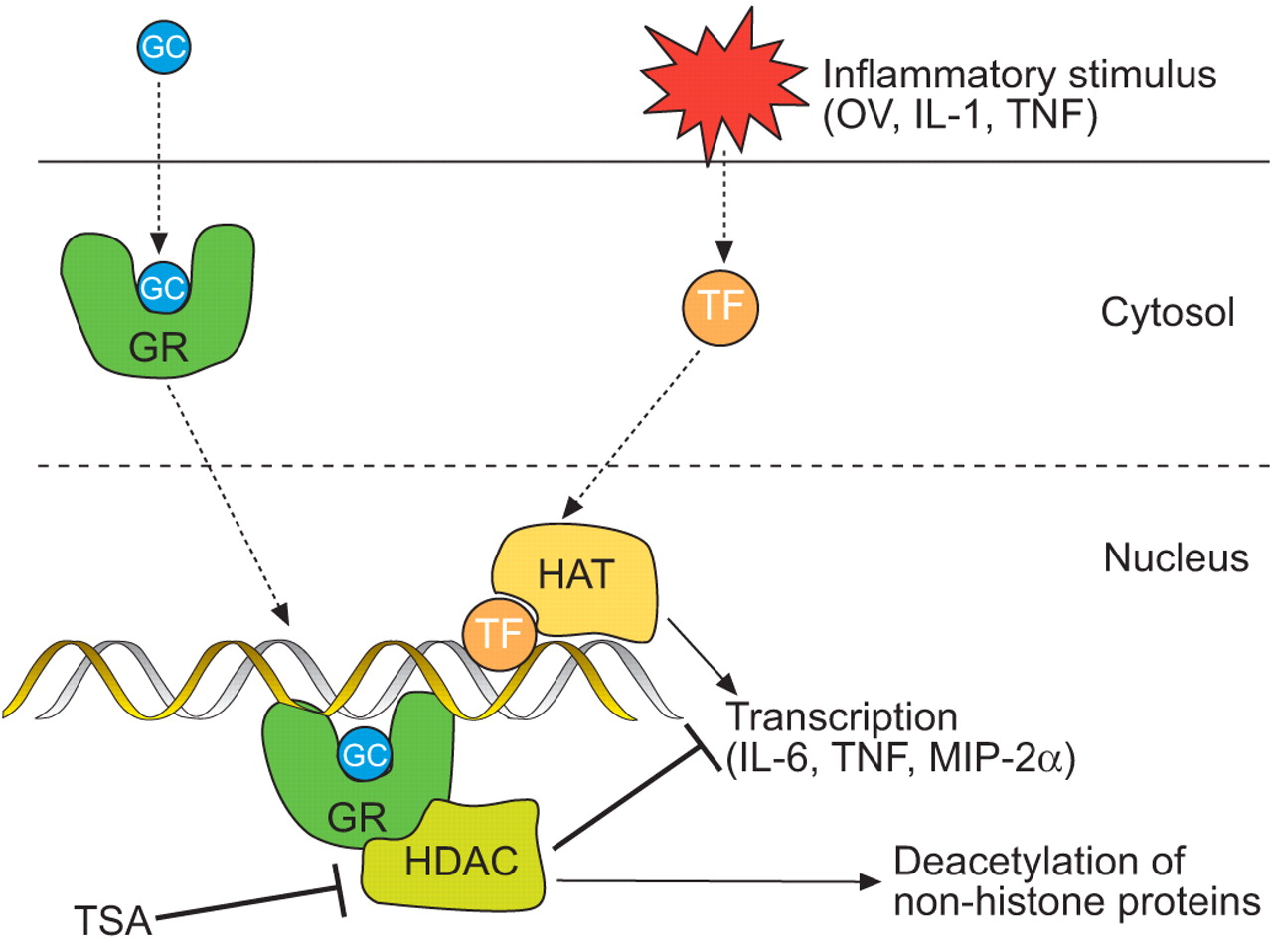
Regulation of histone acetylation offers a novel explanation for the anti-inflammatory action of glucocorticoids (GC) 1, 5. Upon entering the cell, GCs bind to the cytosolic glucocorticoid receptor (GR) and induce its translocation to the nucleus either as a dimer or as a monomer. The dimeric GC/GR complex binds DNA at so-called GC response elements, acting either as a positive (trans-activation, e.g. secretory leukocyte peptidase inhibitor (SLPI) or nuclear factor (NF)-κB inhibitor (IκB)-α) or negative transcription factor (cis-repression) 5. In addition to those mechanisms, it was proposed that steroids repress gene transcription by recruiting HDAC isoenzymes, in particular HDAC2 6, to the activated transcriptional complex where they remove critical acetyl moieties, among others, from the GR 6 and thus bring transcription to an end (fig. 1⇓). This mechanism appears to be especially relevant for NF-κB-dependent genes and low steroid concentrations 6. Steroid-dependent activation of HDAC becomes defective in asthma and COPD and may thus explain the loss of steroid responsiveness in these conditions 1, 5–10.

Hypothetical regulation of inflammatory gene transcription by histone acetylation. Inflammatory stimuli activate transcription factors (TF) that bind to their DNA response elements and recruit histone acetyltransferases (HAT) enabling transcription of cytokines and chemokines by unwinding DNA from histones. Pro-inflammatory gene transcription is partly controlled by glucocorticoid receptors (GR) that translocate to the nucleus and attach to their own DNA binding site. At low glucocorticoid (GC) concentrations, the anti-inflammatory action of steroids is proposed to be mediated by recruitment of histone deacetylases (HDAC), which remove the acetyl moieties and thus reinforce the bond between histones and DNA, hindering transcription of inflammatory mediators. Inhibition of HDAC by trichostatin A (TSA) should thus block the anti-inflammatory effect of glucocorticoids. Besides histones, a number of other proteins are also substrates of the HDAC isoenzymes. OV: overventilation; IL: interleukin; TNF: tumour necrosis factor; MIP: macrophage inflammatory protein.
However, histones are not the only factors of the transcription machinery that are subject to acetylation and HDAC substrates. For instance, regulation by acetylation has been demonstrated for the GR 6 and the transcription factors NF-κB 11–13 and FOX3P 14. Moreover, contrary to the hypothesis discussed previously, it was shown that HDAC inhibitors such as trichostatin A (TSA) are beneficial in models of asthma 15 and rheumatoid arthritis 16, suggesting that TSA with steroids may work in an additive rather than an antagonistic manner.
Thus, depending on the model and possibly on the time course, protein acetylation may be either pro- or anti-inflammatory. A more recently identified stimulus for pro-inflammatory gene induction is ventilatory stress, a phenomenon that is thought to be of great relevance for ventilator-associated lung injury 3, 17. To gain more insight into the regulation of ventilator-induced gene transcription and its modification by steroids, the current authors examined histone acetylation, induction of NF-κB-dependent genes and the effects of steroids under conditions of pulmonary stretch. The study was conducted in isolated perfused mouse lungs, a well-established model for the investigation of stretch-induced gene activation 18–21. Among the advantages of this model are the preserved organ anatomy with full control of the respiratory variables and the lack of extrapulmonary cells that confound interpretation of the results in vivo.
The hypothesis that glucocorticoids act via histone deacetylation leads to the prediction that: 1) ventilation with large tidal volume (VT; overventilation (OV)) enhances histone acetylation; 2) HDAC inhibition augments histone acetylation and inflammatory gene transcription after stimulation by OV; and 3) the steroid dexamethasone (DEX) inhibits inflammatory gene transcription and expression dependent on HDAC activity. The present findings show that the first prediction was true. The second prediction was true for tumour necrosis factor (TNF) and macrophage inflammatory protein (MIP)-2α, but not for interleukin (IL)-6. Prediction three could only partly be confirmed. Thus, regulation of gene transcription by protein acetylation in the lung appears to be specific both for the stimulus and for the gene in question.
MATERIAL AND METHODS
Female BalbC mice (20–25 g) were obtained from FU Berlin (Berlin, Germany) and housed under standard conditions. All experiments were approved by the local authorities.
Isolated perfused mouse lung preparation
Preparation and perfusion (1 mL·min−1 non-recirculating RPMI 1640 medium supplemented with hydroxyethyl starch) of mouse lungs through the pulmonary artery was performed as described previously 18. Briefly, anaesthetised animals were positioned in an open, warmed (37°C) chamber, intubated and ventilated with positive pressure. The abdomen and chest were opened to expose the heart and lungs. The left atrium and pulmonary artery were cannulated and perfusion was started. The chamber was closed airtight and ventilation was switched to negative pressure, in order to guarantee physiological transmural pressure 18. The lungs were perfused and ventilated for 60 min under baseline conditions with an end-inspiratory pressure of -10 cmH2O and an end-expiratory pressure of -3 cmH2O, resulting in a VT of ∼200 µL. VT was measured by numerical integration of airflow velocity. Subsequently, the lungs were randomly allocated to one of the following six groups: 1) low inspiratory pressure of -10 cmH2O (control (C): n = 3); 2) high inspiratory pressure of 22.5 cmH2O (OV: n = 3); 3) high inspiratory pressure as in group 2 plus continuous perfusion with 10−6 M DEX (OV/DEX10−6 M: n = 3); 4) as in group 1 plus continuous perfusion with 100 ng·mL−1 TSA (C/TSA: n = 3); 5) as in group 2 plus TSA (OV/TSA: n = 3); and 6) as in group 3 plus TSA (OV/DEX10−6 M /TSA: n = 3). The experiments were carried out for 180 min after randomisation. In a second set of experiments, lungs were perfused exactly as above, but randomly allocated to one of four groups: 1) C (n = 6); 2) OV (n = 5); 3) OV plus 10−8 M DEX (OV/DEX10−8 M: n = 7); and 4) OV plus 10−8 M DEX plus 100 ng·mL−1 TSA (OV/DEX10−8 M/TSA: n = 7). Data from both sets of experiments were combined for ELISA and relative quantification (RQ)-PCR.
After passage through the lung, perfusate was sampled every 30 min and frozen immediately in liquid nitrogen. At the end of the experiment, the lung was disconnected, excised from the corpse and frozen in liquid nitrogen. All samples were kept at -80°C.
Lungs were ground up in liquid nitrogen using a pestle and mortar. For preparation of histone extracts and nuclear extracts, lung powder was thawed and washed with cold PBS.
Apoptosis assay
For detection of caspase-8 activity as an early marker of apoptosis, a kit was purchased from BD Biosciences (Palo Alto, CA, USA) and the assay was performed according to the manufacturer’s instructions. Briefly, lung powder was washed in PBS, lysed and centrifuged. Protein content was determined from an aliquot of the supernatant and another aliquot was incubated with reaction buffer and caspase-8 substrate for 3 h at 37°C. The absorbance was measured at 405 nm and normalised to protein content. As a positive control, human microvascular endothelial cells (HMVEC) cells were subjected to gamma irradiation (100 Gy) and subsequently incubated at 37°C for 4 h to enhance the signal.
Preparation and immunoblotting of nuclear extracts
Nuclei were extracted according to the method of Dignam et al. 22. Briefly, lung powder was lysed in cell lysis buffer (10 mM Tris/HCl, pH 7.9, 1 mM EGTA, 10 mM KCl; Complete Mini (Roche Diagnostics GmbH, Mannheim, Germany), 0.6% NP-40). Nuclei were pelleted and incubated with nuclear extraction buffer on ice (20 mM Tris/HCl, pH 7.9, 1 mM EGTA, 420 mM NaCl, 25% glycerol; Complete Mini). After centrifugation, the supernatant containing nuclear proteins was collected and stored at -80°C.
Equal amounts of protein were separated by sodium dodecyl sulphate (SDS)-polyacrylamide gel electrophoresis and transferred to nitrocellulose. Membranes were incubated with antibodies against HDAC1–3 (Upstate, Dundee, UK and Santa Cruz Biotechnology, Santa Cruz, CA, USA) and subsequently with fluorescent labelled secondary antibodies (Invitrogen GmbH, Karlsruhe, Germany and Rockland Immunochemicals, Gilbertsville, PA, USA). HDACs were detected on a LI-COR Odyssey Imaging System (LI-COR Biosciences, Bad Homburg, Germany).
Preparation of histone extracts
Histones were extracted from lung powder as described previously 23 with slight modifications. Briefly, lung powder was washed in PBS and centrifuged. The pellet was suspended in lysis buffer (10 mM Tris/HCl, pH 6.5, 50 mM Na bisulphite, 10 mM MgCl2, 8.6% sucrose, 0.5% NP-40, 10 mM Na-butyrate; Complete Mini) and incubated for 10 min on ice. After centrifugation (13,500×g, 4°C), the pellet was washed three times with lysis buffer and once with 10 mM Tris/HCl, pH 7.4, 13 mM EDTA. The pellet was then suspended in 0.2 M H2SO4 and incubated on ice for 1 h with occasional mixing. The dissolved histones were separated by centrifugation (13,500×g, 4°C) and precipitated with acetone at -20°C overnight. The precipitate was centrifuged and washed with ice-cold acetone. The remaining pellet was air dried on ice and dissolved in 50 mM Tris/HCl, pH 7.9, 10mM NaCl, 0.25 mM EDTA, 10 mM 2-mercaptoethanol, 10% glycerol. Protein concentration was determined and the histone solution kept at -80°C.
Immunoblotting of histones
Histones were separated according to their size, charge and hydrophobicity on 12% AUT gels. Briefly, histone extracts were mixed with the same volume of sample buffer (8 M urea, 0.7 mM 2-mercaptoethanol, 20% glycerol, 6% (volume/volume) acetic acid, pyronin Y) and incubated at 50°C for 5 min. Volumes equivalent to 10–20 µg of protein were loaded onto the gel and separated in running buffer (100 mM glycine, 0.6% acetic acid) with inverted current overnight. Gels were then equilibrated in transfer buffer (25 mM Tris/HCl, pH 8.3, 192 mM glycine, 0.1% SDS) and transferred to nitrocellulose. Transfer was checked with Ponceau S dye. The membrane was blocked with RotiBlock (Roth, Karlsruhe, Germany) and incubated with antibodies against differently acetylated histones H3 (Santa Cruz Biotechnology) and H4 (Upstate-Millipore, Billerica, MA, USA). Fluorescently labelled secondary antibodies were used (Invitrogen GmbH and Rockland) and acetylated histones detected on LI-COR Odyssey Imaging System (LI-COR Biosciences). In H4, the signal of all bands was added up to total H4 fluorescence. In H3, the band representing monoacetylated protein was neglected when adding up fluorescence, as it was the biggest band and changes were rather small.
Cytokine concentration in the perfusate
For detection of TNF-α, IL-6 and MIP-2α in perfusate samples, commercially available tests (OptEIATM Set Mouse TNF-α (mono/poly) and OptEIATM Mouse IL-6 Set; BD Biosciences, San Diego, CA, USA, and Quantikine Mouse MIP-2 Immunoassay; R&D Systems GmbH, Wiesbaden, Germany) were purchased and performed exactly according to the manufacturers' instructions.
Real-time RQ-PCR
For isolation of mRNA, an RNA-isolation kit was purchased from Roche (Mannheim, Germany). Lung powder was suspended in GUTC buffer (4 M guanidinium thiocyanate, 0.5% N-lauroylsarcosine, 25 mM Na-citrate, 1% 2-mercaptoethanol). Following this, 400 µL lysis buffer was added and DNA sheared by passing the lysate through a 20-gauge needle. After centrifugation, the supernatant was mixed with 200 µL ethanol and applied to the column. Columns were treated with DNAse, to remove all adhering genomic DNA, and then washed. RNA was eluted and subjected to reverse transcription.
For real-time RQ-PCR, specific primers were chosen from the literature and primer properties tested using Omiga 2.0 Software (Oxford Molecular, Oxford, UK). Primer sequences are as follows. TNF-α: sense 5′-TCT-CAT-CAG-TTC-TAT-GGC-CC-3′, antisense 5′-GGG-ATG-AGA-CAA-GGT-ACA-AC-3′; IL-6: sense 5′-CCA-GAG-ATA-CAA-AGA-AAT-GAT-GG-3′, antisense 5′ACT-CCA-GAA-GAC-AGG-AAA-T-3′; MIP-2α: sense 5′-AGT-GAA-CTG-TGT-CAA-TGC-3′, antisense 5′-AGG-CAA-ACT-TTT-TGA-CCG-CC-3′; HDAC1: sense 5′-AAT-TCC-TGC-GTT-CTA-TTC-G-3′, antisense 5′-CAA-ACA-AGC-CAT-CAA-ATA-CC-3′; HDAC2: sense 5′-GTT-CAA-ATG-CAA-GCT-ATT-CC-3′, antisense 5′-ACC-TCC-TTC-ACC-TTC-ATC-C-3′; and β2-microglobulin: sense 5′-TGA-CCG-GCT-TGT-ATG-CTA-TC-3′, antisense 5′-CAG-TGT-GAG-CCA-GGA-TAT-AG-3′. PCR amplification was performed in triplicate. Gene expression was normalised to an endogenous reference gene, mouse β2-microglobulin.
Statistics
Data are presented as mean±sem. Statistical comparisons were performed with JMP 5.0.1 for Windows and R 2.0 24. Data for figures 2⇓–⇓⇓⇓6⇓ were transformed by the Box-Cox transformation and subjected to univariate ANOVA. For comparison of VT curves, the current authors used the area under the curve from 60–240 min (Prism 4.03; GraphPad Software, Inc., San Diego, CA, USA). In figures 7⇓ and 8⇓ each control represents exactly 100% (as there was always only one control per gel); ANOVA was not applicable so the nonparametric Mann–Whitney rank test was performed. Seven comparisons were made: C versus C/TSA; C versus OV; OV versus OV/TSA; OV versus OV/DEX10−6 M; OV versus OV/DEX10−8 M; OV/DEX10−6 M versus OV/DEX10−6 M/TSA; and OV/DEX10−8 M versus OV/DEX10−8 M/TSA. Multiple comparisons were accounted for by the false discovery rate for all figures.

Tidal volumes (VT) during ventilation of isolated perfused mouse lungs. After ventilation with -3/-10 cmH2O (end-expiratory/end-inspiratory pressure) for 1 h, lungs were ventilated with low pressure (C) or high pressure (-22.5 cmH2O end-expiratory pressure; OV) for 3 h and perfused with trichostatin A (TSA), dexamethasone (DEX) or both. a) VT and b) comparison of VT. The area under the curve (AUC) was calculated from 60–240 min. ○: control; •: C/TSA; □: OV; ▪: OV/TSA; ▵: OV/DEX 10−6M; ▴: OV/DEX 10−6M/TSA; ⋄: OV/DEX 10−8M; ♦: OV/DEX 10−8 M/TSA. Data are shown as mean±sem from n = 3–9 to independent experiments each. ns: not significant. ***: p<0.001.

Caspase-8 as an early marker of apoptosis. After ventilation with -3/ -10 cmH2O end-expiratory/end-inspiratory pressure for 1 h, lungs were ventilated with low pressure (C) or high pressure (-22.5 cmH2O end-expiratory pressure; OV) for 3 h and perfused with trichostatin A (TSA), dexamethasone (DEX) or both. At the end of the experiment, lungs were frozen in liquid nitrogen, ground to a powder and analysed for caspase-8 activity. As a positive control, human microvascular endothelial cells were subjected to gamma irradiation (100 Gy) and cultivated for 4 h at 37°C for maximal caspase-8 activity. AU: arbitrary units; APC: apoptosis control. ········: assay background.

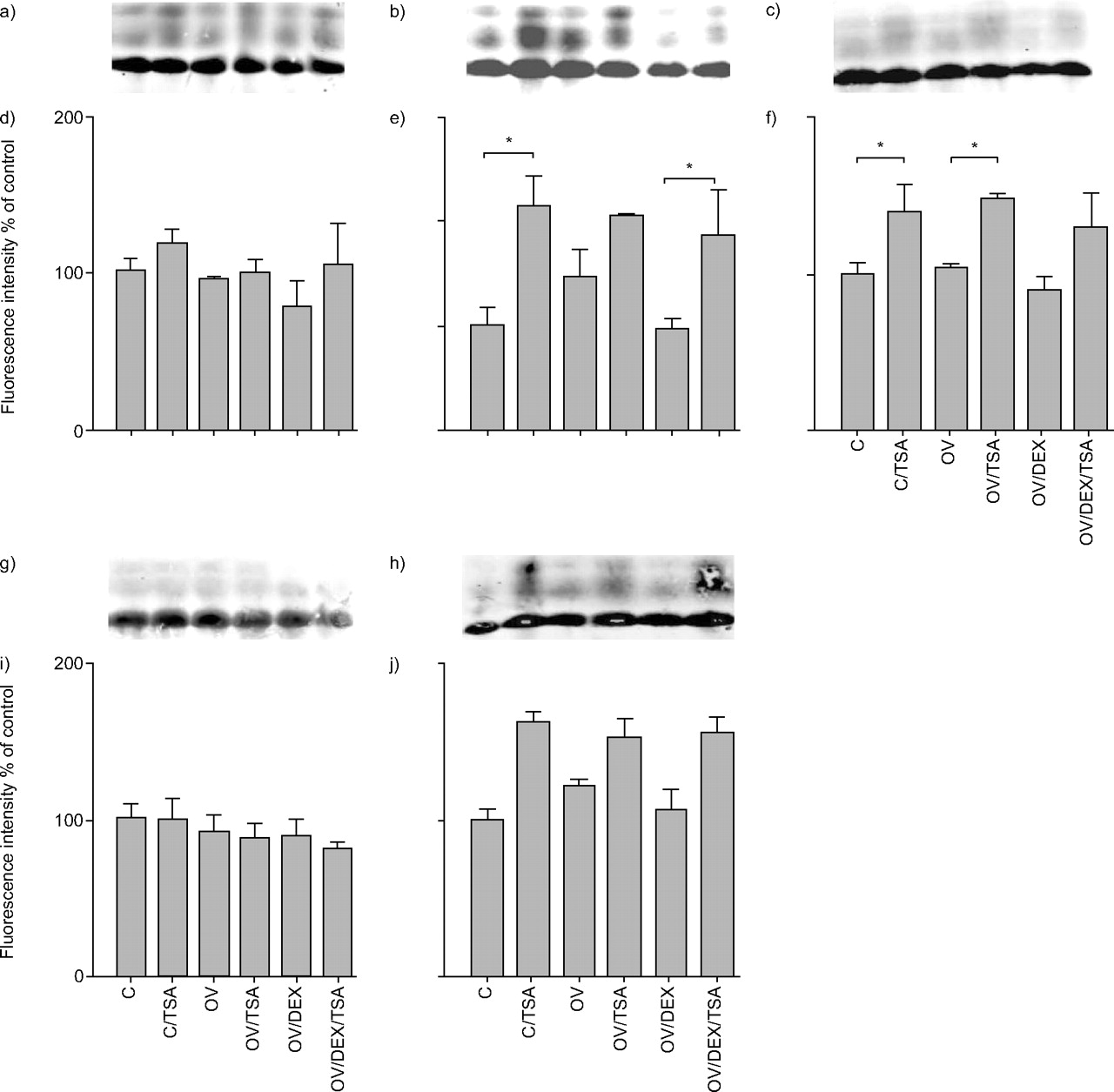
Expression of histone H3 acetylation; a and d) H3 lysine (K)9; b and e) H3K14; c and f) H3K18; g and i) H3K23; h and j) H3K27. a, b, c, g and i) Histone extracts were probed by immunoblotting with antibodies against different acetylated K residues and visualised by fluorescence-labelled secondary antibodies. After ventilation with -3/-10 cmH2O (end-expiratory/end-inspiratory pressure) for 1 h, lungs were ventilated with low pressure (C) or high pressure (-22.5 cmH2O end-expiratory pressure; OV) for 3 h and perfused with trichostatin A (TSA), dexamethasone (DEX) or both. At the end of the experiment, lungs were frozen in liquid nitrogen. n = 3–4. *: p<0.05.

Transcription of a) histone deacetylases (HDAC)1 mRNA levels and b) HDAC2 mRNA levels. After ventilation with -3/-10 cmH2O (end-expiratory/end-inspiratory pressure) for 1 h, lungs were ventilated with low pressure (C) or high pressure (-22.5 cmH2O end-expiratory pressure; OV) and perfused with trichostatin A (TSA) or dexamethasone (DEX) or both. After 3 h, perfusate samples were collected and lungs frozen in liquid nitrogen. RNA was prepared from lung powder and subjected to reverse transcription and relative quantification-PCR. mRNA levels were normalised to β2-microglobulin and expressed relative to control (C) levels. n = 3. *: p<0.05; **p<0.01; ***: p<0.001.
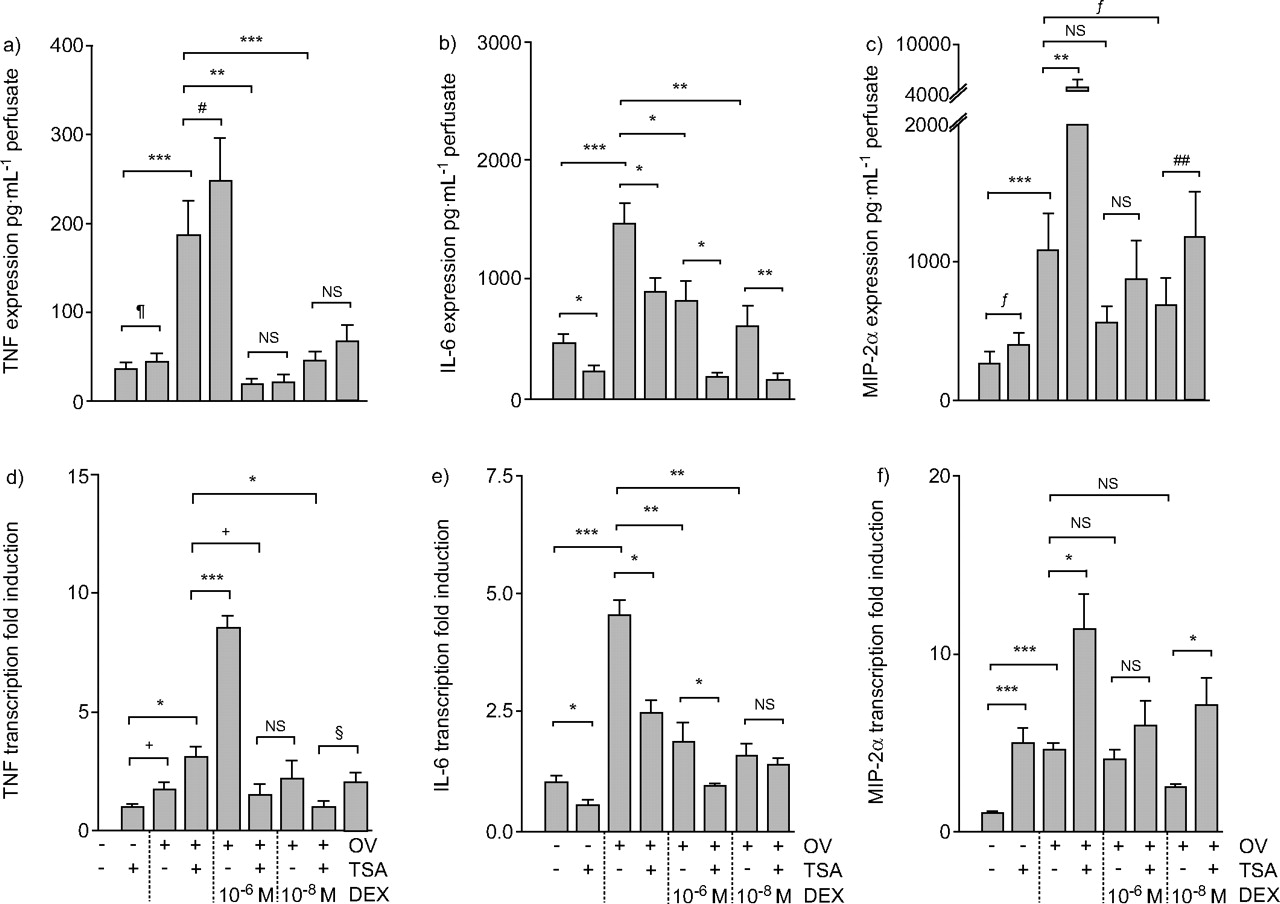
Cytokine perfusate levels and tissue mRNA levels. Tumour necrosis factor (TNF) expression (a), TNF transcription (d), interleukin (IL)-6 expression (b), IL-6 transcription (e), macrophage inflammatory protein (MIP)-2α expression (c) and MIP-2α transcription (f). After ventilation with -3/-10 cmH2O (end-expiratory/end-inspiratory pressure) for 1 h, lungs were ventilated with low pressure or high pressure (-22.5 cmH2O end-expiratory pressure; OV) and perfused with trichostatin A (TSA), dexamethasone (DEX) or both. After 3 h, perfusate samples were collected and lungs frozen in liquid nitrogen. TNF and MIP-2α perfusate concentration were measured by ELISA. RNA was prepared from lung powder and subjected to reverse transcription and relative quantificaion-PCR. mRNA levels were normalised to β2-microglobulin and expressed relative to control levels. n = 3–9. ns: not significant. #: p = 0.090; ¶: p = 0.164; +: p = 0.199; §: p = 0.146; ƒ: p = 0.122; ##: p = 0.159. *: p<0.05; **p<0.01; ***: p<0.001.
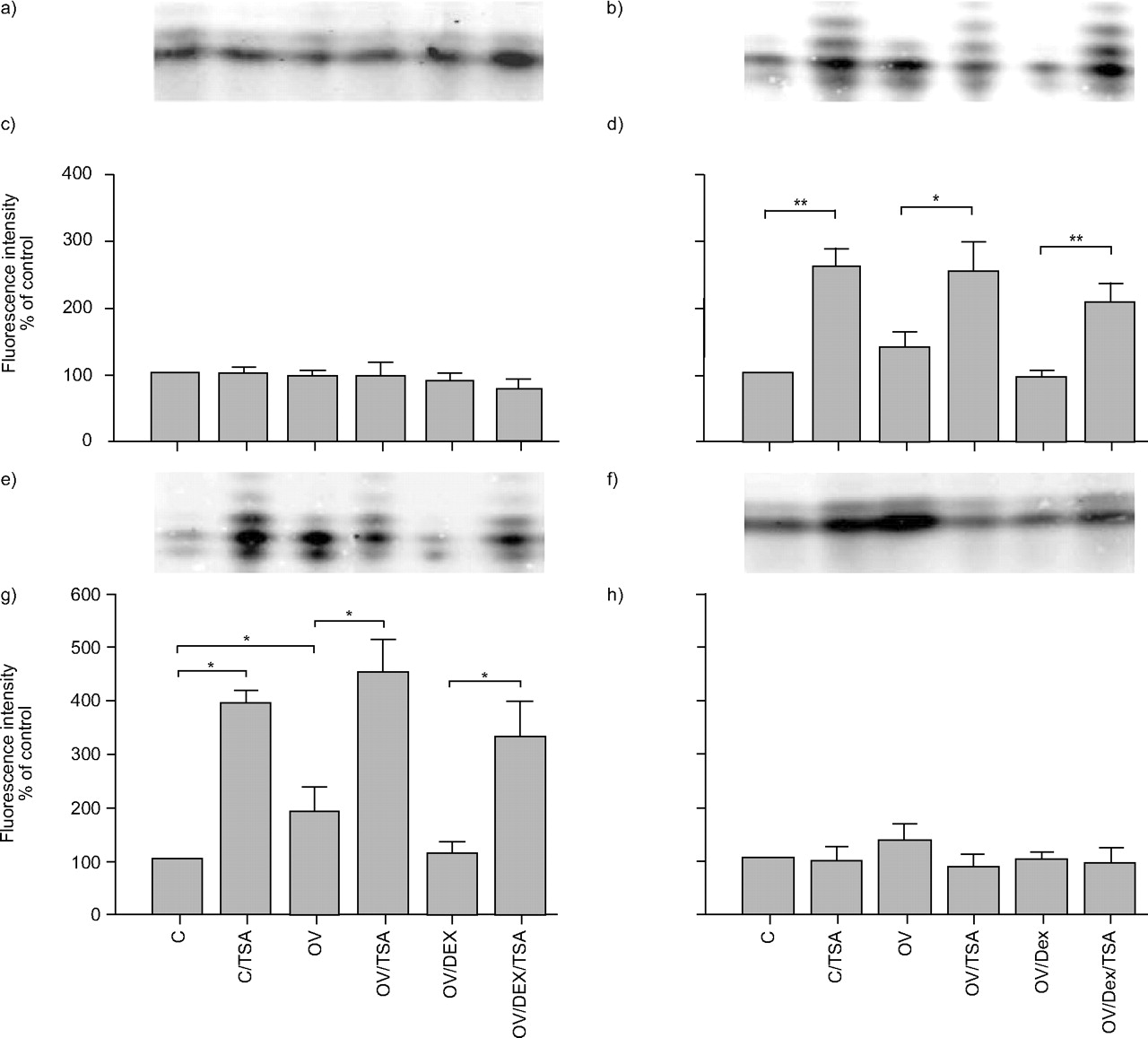
Expression of histone H4 acetylation: a and c) H4 lysine (K)5; b and d) H4K8; e and g) H4K12; f and h) H4K16. a, c, e and f) Histone extracts were probed by immunoblotting with antibodies against different acetylated K residues and visualised by fluorescence-labelled secondary antibodies. After ventilation with -3/-10 cmH2O (end-expiratory/end-inspiratory pressure) for 1 h, lungs were ventilated with low pressure (C) or high pressure (-22.5 cmH2O end-expiratory pressure; OV) for 3 h and perfused with trichostatin A (TSA) or dexamethasone (DEX) or both. At the end of the experiment, lungs were frozen in liquid nitrogen. n = 5–6. *: p<0.05; **: p<0.01.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Expression of a) histone deacetylase (HDAC)1, b) HDAC2 and c) HDAC3. After ventilation with -3/-10 cmH2O (end-expiratory/end-inspiratory pressure) for 1 h, lungs were ventilated with low pressure (C) or high pressure (-22.5 cmH2O end-expiratory pressure; OV) for 3 h and perfused with trichostatin A (TSA), dexamethasone (DEX) or both. At the end of the experiment, lungs were frozen in liquid nitrogen. Nuclear extract proteins were separated by sodium dodecyl sulphate-polyacrylaminde gel electrophoresis and transferred to nitrocellulose. HDACs were stained with specific antibodies and visualised by fluorescence labelled secondary antibodies. n = 6. *: p<0.05.
RESULTS
VT
Control lungs ventilated with -3/-10 cmH2O end-inspiratory/end-expiratory pressure had a VT of 0.28±0.01 mL and 0.25±0.01 mL after 60 and 240 min, respectively. The VT of lungs ventilated with -3/-22.5 cmH2O rose to 0.68±0.02 mL after application of OV, and slowly declined thereafter (fig. 2a⇑). Neither TSA nor DEX significantly affected the VT in this model (fig. 2b⇑).
Caspase activity
As TSA induces cell cycle arrest and apoptosis in carcinoma cell lines 25, caspase-8 activity was determined as one of the earliest events within the apoptosis cascade, in order to rule out any adverse effect of the HDAC inhibitor on the isolated perfused mouse lungs. As a positive control, the present authors used HMVEC subjected to gamma irradiation. HMVECs, but none of the experimental groups, showed caspase-8 activity above the assay background (fig. 3⇑).
Histone acetylation
The current authors examined lysines (K)-5, K8, K12, and K16 in histone H4, and K9, K14, K18, K23, and K27 in histone H3. In histone H4, up to five equidistant bands corresponding to mono-, di-, tri- and tetra-acetylated histone were detected, plus a so far unknown band possibly resulting from another charged (phosphate) or charge-neutralising (acetyl) protein modification. In controls stained for K5 or K16 acetylation, most fluorescence was detected within one band, much of it probably corresponding to monoacetylated histone H4 (fig. 7a⇑ and f). A small signal was seen in the band attributed to diacetylated histone H4K5. OV did not increase the level of acetylation of K5 or K16, and neither did TSA or DEX.
In controls stained for acetylated histone H4K8 (fig. 7b⇑) or H4K12 (fig. 7e⇑), three distinct bands were clearly visible, the strongest being the middle one. TSA increased acetylation of H4K8 and H4K12 under all experimental conditions to between 250 and 500% of control. H4K8 showed two more bands above the other three, corresponding to a reduced positive charge consistent with lysine acetylation. This site tended towards higher levels of acetylation in OV lungs (p = 0.06). Acetylation of K12 was increased by OV (fig. 7g⇑) and this was reversed by DEX.
In histone H3, up to four equidistant bands were detectable (fig. 4a–c⇑, g and h), the lowest being the most intense and probably consisting of monoacetylated histone H3. Acetylation triggered by TSA was less pronounced in histone H3 than in histone H4. To be able to detect these minute differences, for quantification the lowest band was excluded. Acetylation of K9, K23 and K27 did not respond to TSA (p>0.05; fig. 4d⇑, i and j). In K14 and K18, up to 114% more di- and tri-acetylated protein was found in response to TSA, irrespective of the mode of ventilation or administration of DEX (fig. 4e⇑ and f). OV did not increase acetylation of any lysine residue in H3.
HDAC protein expression and transcription
To exclude the possibility that alterations in HDAC protein expression explain the increased histone acetylation, immunoblots were performed. The current authors found no major differences in the expression of HDAC1, HDAC2 or HDAC3 protein under all experimental conditions (fig. 8a–c⇑). TSA tended to decrease total HDAC enzymatic activity under all experimental conditions; however, consistent measurement of HDAC activity proved difficult in whole lung extracts (data not shown).
RQ-PCR for HDAC1 and HDAC2 after isolated lung perfusion was also performed (fig. 5a⇑ and b). Here, the present authors found higher levels of HDAC1 and HDAC2 mRNA in all groups treated with TSA. Remarkably, there was a 50% reduction in the HDAC2 mRNA level in response to OV (fig. 5b⇑), which was largely prevented by DEX treatment. These alterations in gene expression were not reflected at the protein level.
Gene expression and cytokine production
An important part of the present study was to examine the effect of TSA on OV-induced production of MIP-2α, TNF and IL-6; NF-κB-dependent genes that are known to become activated in overventilated mouse lungs 18, 20. As histone acetylation promotes gene transcription, it was hypothesised that inhibiting HDACs by TSA would increase the expression of inflammatory mediators. As cytokine production is frequently subject to post-transcriptional control, both pulmonary gene transcription and pulmonary cytokine production (perfusate levels) were examined. In addition, because the effect of steroids on HDAC was suggested to be relatively more pronounced at lower steroid concentrations 5, DEX was examined at a low (10−8 M) and a high (10−6 M) concentration. Since TSA had fundamentally different effects on MIP-2α and TNF on the one hand, and on IL-6 on the other hand, the corresponding data will be presented separately.
TNF and MIP-2α
In control lungs, treatment with TSA increased MIP-2α gene expression (fig. 6f⇑) but had no effect on MIP-2α production (fig. 6c⇑), TNF gene expression (fig. 6d⇑) or TNF production (fig. 6a⇑).
As expected, OV increased gene expression and production of MIP-2α and TNF. In the case of MIP-2α, this effect was strongly increased by TSA (fig. 6c⇑ and f); in the case of TNF, the increase induced by TSA was significant for gene expression (fig. 6d⇑) but not for cytokine production (p = 0.09; fig. 6a⇑).
In agreement with previous findings 19, MIP-2α production was alleviated but not prevented by DEX (fig. 6c⇑); MIP-2α gene expression was essentially unaltered (fig. 6f⇑). TSA treatment tended to increase MIP-2α transcription (fig. 6f⇑) and MIP-2α production in OV/DEX treated lungs (fig. 6e⇑); this was significant for gene expression in lungs treated with OV and 10−8 M DEX (fig. 6f⇑). OV-induced TNF gene transcription was reduced by 10−8 M DEX (fig. 6d⇑) and TNF production was reduced by both concentrations of DEX (fig. 6a⇑). TSA tended to attenuate the DEX-mediated reduction in OV-induced TNF transcription and production, but this effect was always minor and not significant (fig. 6a⇑ and d⇑).
IL-6
TSA treatment reduced IL-6 gene expression (fig. 6e⇑) and production (fig. 6b⇑) in control lungs. As expected, OV increased both IL-6 gene expression (fig. 6e⇑) and IL-6 production (fig. 6b⇑). This effect was largely blocked by TSA treatment (fig. 6c⇑ and d). DEX prevented OV-induced IL-6 production, an effect that was further amplified by TSA (fig. 6b⇑ and e).
DISCUSSION
Histone acetylation is a central mechanism of gene regulation as it relieves DNA from core histones and thus enables the transcriptional machinery to bind and transcribe DNA. Recently, the role of histone acetylation in inflammatory gene transcription is receiving increasing attention, which is fuelled by the exciting notion that alterations in this key regulatory mechanism may help to explain steroid resistance in difficult-to-treat conditions, such as steroid-resistant asthma or COPD. Another inflammatory disorder associated with oxidative stress where steroids are surprisingly ineffective is acute respiratory distress syndrome (ARDS) 26. Since mechanical ventilation is mandatory in ARDS and has immense impact on outcome 27, it is of considerable interest to know how mechanical ventilation affects histone acetylation and to what extent the anti-inflammatory actions of steroids depend on histone deacetylation under these conditions. Therefore, the current authors examined histone acetylation in intact mouse lungs subjected to high constant-pressure ventilation (OV). The present findings show that OV activates histone acetylation. Particularly noteworthy is the observation that inhibition of HDAC activity with TSA increased transcription of TNF and MIP-2α, but reduced that of IL-6. With respect to the effects of DEX, which decreased OV-induced cytokine expression, TSA partly reversed its effect on MIP-2α transcription but further enhanced the decrease in IL-6. These findings show that the regulation of gene transcription by HDACs is gene dependent and complex.
Technical comments
The current experiments were performed in isolated perfused mouse lungs, a model that permits the detailed analysis of ventilator-induced activation of pro-inflammatory genes 20. It should be noted, however, that this is not a model of ventilator-induced lung injury, as during OV the lung structure remains largely intact 18. In fact, the argument can be made that an intact pulmonary microanatomy is a prerequisite for (patho)physiological mechanotransduction processes 28. Nonetheless, it is clear that ventilator-induced cytokine production is a critical determinant of ventilator-induced lung injury 3, 17, 29.
TSA has been used as a HDAC inhibitor in a number of studies. Originally described in 1976 as a new antifungal antibiotic 30, it was found to induce differentiation 31 and cell cycle arrest 32. In 1990, Yoshida et al. 33 proposed that TSA induces histone hyperacetylation by inhibition of HDACs at nanomolar concentrations. All subsequent studies with TSA reproduced the histone hyperacetylation 34–36 and the inhibition of HDAC 37–39. The observation that TSA fits in the catalytic pocket of an HDAC-like protein together with the conservation of active site and contact residues across the HDAC family 40 finally established TSA as an HDAC inhibitor.
Thus, TSA appears to be a fairly specific HDAC inhibitor. However, because HDAC isoenzymes have substrates other than histones, such as the GR 6, NF-κB 11–13, FOXP3 14 or microtubuli 41, and interact with protein phosphatases 41, the effects of TSA must not be explained by alterations in histone acetylation alone. It remains to be shown whether other effects of TSA that have been described, such as increased activity of PI-3-kinase 42, 43 or phosphorylation of the epidermal growth factor-receptor 43, are also mediated by HDAC isoenzymes or are unspecific side-effects of TSA. Thus, TSA is a tool for probing HDAC activity but cannot be used to specifically address histone acetylation.
As TSA had been shown to induce apoptosis in various cancer cell lines, apoptosis was checked for at the end of the experiments. Since caspase-8 activity was not detected (as one of the earliest markers of apoptosis) in any experimental group, it was concluded that TSA did not induce apoptosis in the current model.
After treatment with TSA, the present authors detected five equidistant bands of acetylated histone H4, which, to the best of their knowledge, has not been observed before. AUT gels separate proteins not only by size but also by hydrophobicity and, importantly, by charge. This implies the existence of at least five differently charged and, therefore, modified histones that all carry at least one acetyl group. Even though, theoretically, H4 can be acetylated at up to 15 sites, only four acetylation sites (K5, K8, K12 and K16) have so far been identified and studied in detail 44. Apart from the existence of a so far unknown acetylation site, phosphorylation at H4 serine 1 has been described 45 and alters the charge of the protein by two units but has been linked with hypoacetylation of H4 in yeast 46. Other modifications, such as methylation, SUMOylation and ubiquitination, can also occur but do not introduce a different charge or alter the size of the histone so substantially that major changes would be expected in the electrophoretic mobility. Thus, acetylation seems to be the most likely modification within these equidistant bands. The appearance of two bands with lower positive charge after treatment with the HDAC inhibitor TSA also points towards acetylation. However, one or two of the more positively charged bands might be phosphorylated as well as acetylated under basal conditions.
Hypothesis 1: ventilation with large VT enhances histone acetylation
As expected, acetylation and deacetylation at specific lysine residues occurred even under baseline conditions. Perfusion of intact mouse lungs with the HDAC inhibitor TSA resulted in hyperacetylation of H4K8 and H4K12, whereas H4K5 and H4K16 remained unaffected. At histone H3, small increases in K14 and K18 acetylation were found after TSA treatment, while K9, K23 and K27 acetylation remained at basal levels. These results once again show the specificity of histone modifications and further demonstrate the possibility of analysing histone acetylation within the intact organ.
OV resulted in acetylation of H4K12 and possibly H4K8, which was reversed in both instances by treatment with the glucocorticoid DEX. High-volume ventilation is a pro-inflammatory stimulus prompting the secretion of cytokines and chemokines via activation of NF-κB 3, 19 and activator protein-1 47–49 and thus behaves like other inflammatory stimuli, such as TNF, IL-1β, H2O2 or cigarette smoke that act via similar pathways. Remarkably, in A549 cells these stimuli produced a histone acetylation pattern 50–55 comparable to OV, extending the similarities between classical pro-inflammatory stimuli and OV to alterations in histone acetylation. Also in line with the present data, the upregulation of H4K8 and H4K12 acetylation in A549 cells after stimulation with IL-1β was prevented by treatment with 10−6 M DEX 52. Taken together, these findings suggest that the H4K8 and H4K12 histone acetylation sites are particularly relevant for inflammatory gene transcription. Furthermore, acetylation of these sites appears to be partly regulated by steroids, also in the case of OV.
Hypothesis 2: inhibition of HDACs augments histone acetylation and inflammatory gene transcription
According to the model developed by Barnes and co-workers 1, 5, inhibition of HDAC is expected to increase pro-inflammatory gene expression. The present findings show that this is true for some but not all pro-inflammatory genes, at least if the stimulus is mechanical stretch. On the one hand, TSA enhanced the transcription and production of MIP-2α and TNF in response to high-volume ventilation. On the other hand, however, IL-6 transcription and production was reduced by TSA. This latter observation is in line with the effect of TSA on IL-4, IL-5 and immunoglobulin E in a mouse asthma model 15. Other authors have also reported that HDAC inhibitors may attenuate the expression of various genes 56–58. To further complicate matters, TNF, IL-6 and MIP-2α/IL-8 have been found to be enhanced by TSA in some cells types but reduced in others 54, 56, 58–60.
A number of explanations for these phenomena seem possible. First, the classical view is that deacetylation of histones represses transcription. In keeping with this, HDAC inhibition further activated pre-stimulated genes explaining the synergistic effect of OV and TSA on TNF and MIP-2α expression in the current model. However, as discussed previously, HDACs also acetylate a number of non-histone proteins and thus regulate their activity. As noted above, NF-κB, a pro-inflammatory transcription factor activated by OV 19, is subject to regulatory acetylation. For instance, acetylation of the Rel A/p65 subunit of NF-κB (by CBP/p300) increases its DNA binding affinity, whereas deacetylation by HDAC3 promotes binding to IκB-α and rapid export from the nucleus 11. Like altered histone acetylation, such a mechanism would explain the increased expression of TNF and MIP-2α by TSA in the present model.
Conversely, other studies observed inhibition of NF-κB nuclear transportation and p50 dimer activity by the HDAC inhibitor butyrate 61, 62, pointing towards the opposite direction, which would be one possibility to explain the findings on IL-6. Acetylation of non-histone proteins was also put forward as an explanation for the repression of the mouse mammary tumour virus by TSA in the presence and absence of glucocorticoids 63. Furthermore, the alterations in gene expression induced by TSA may change with time. In stimulated N9 microglial cells, TSA inhibited IL-6 secretion for up to 12 h but increased it at later time points 64. Another alternative is that HDACs might regulate inducers or repressors of cytokine transcription.
The antithetic cytokine answers to TSA indicate that the genes that are turned on by OV are regulated by different mechanisms, which is in line with previous conclusions 65. TNF, which was not enhanced by TSA in control lungs, might be under tight control with absolute necessity of transcription factor binding. MIP-2α transcription is augmented by TSA and thus probably pre-stimulated under control conditions where transcription is hindered only by the predominance of HDACs over HATs. IL-6 is easily induced by OV but, unlike TNF and MIP-2α, repressed by TSA. IL-6 expression may thus be under the control of an unknown repressor whose transcription is initiated by altering the balance between HAT and HDAC.
Hypothesis 3: DEX inhibits inflammatory gene transcription and expression dependent on HDAC activity
More recently it was suggested that HDAC2-dependent deacetylation of the GR is particularly relevant to explain the attenuation of NF-κB-dependent gene expression by low steroid concentrations 6. TNF, IL-6 and MIP-2α are all NF-κB-dependent genes, and apparently regulated through this pathway during OV 19, 21. However, in the present model, TSA tended to reverse only the steroid-induced reduction in MIP-2α and TNF production (this was statistically significant only for the transcription of MIP-2α at low steroid concentrations). These findings do support, at the whole organ level, the hypothesis that the anti-inflammatory action of steroids is partly mediated by HDAC activity, at least with respect to ventilation-induced expression of TNF and MIP-2α. Yet it also became apparent that even for these two cytokines, a large and significant part of the anti-inflammatory action of steroids must be mediated by HDAC-independent mechanisms. In addition to mechanisms discussed previously, one alternative mechanism of how steroids may downregulate expression of pro-inflammatory proteins is destabilisation of their mRNA, as was shown for cyclooxgenase-2 66. This mechanism is mediated by an adenine-uracil-rich element in the 3′-untranslated region of the gene 67, which is also present in the mRNA of TNF and MIP-2α.
As already noted above, the mechanism of steroid action is most likely gene- and cell-dependent. Thus, the fact that the articles establishing the link between glucocorticoids and HDACs mostly looked at the expression of granulocyte-macrophage colony-stimulating factor and SLPI 6, 52, 68, 69, which were not readily induced by OV (data not shown), while the current authors examined TNF, MIP-2α and IL-6, might account for some of the differences between these studies, in particular for the decrease in IL-6 expression and for the much smaller role of HDAC in explaining the anti-inflammatory effects of steroids.
Clinical implications
First, with respect to inflammatory lung disease, both restoration (by theophylline) 70 and inhibition of HDAC have been suggested as possible treatment options 15. This discrepancy is in keeping with one result of the present study: namely that, depending on the gene, altering HDAC activity may either intensify (TNF, MIP-2α) or reduce (IL-6) the expression of inflammatory genes. Thus, the treatment of inflammatory disorders with the aim of altering HDAC activity may produce complex results and should be viewed with caution.
Secondly, the decrease in HDAC2 transcription by 50% caused by OV is of potential clinical relevance. Even though at the end of the experiment the current authors did not find a similar response in HDAC protein expression, this might be due to the time-frame of the isolated lung perfusion. In the intact unstimulated lungs the level of HDAC protein is kept constant by concurrent gene expression and protein degradation. If both processes are relatively slow, a decrease in protein levels in response to declining gene expression might not be detectable for some time and thus exceed the viability of the isolated organ. Taking into account the importance of histone deacetylation in regulating the inflammatory response, a lack of HDAC2 might alter inflammatory responses during mechanical ventilation at later time-points. However, this needs to be addressed in further studies.
CONCLUSION
In summary, the current authors demonstrated that: 1) studying histone acetylation and deacetylation is feasible in the intact lung; 2) high-volume ventilation enhances histone acetylation at specific sites; 3) inhibition of histone deacetylases augments histone acetylation and has differential effects on the expression of inflammatory genes, enhancing some (tumour necrosis factor, macrophage inflammatory protein-2) and decreasing others (interleukin-6); and 4) steroids modify overventilation-induced gene activation probably partly, but by no means exclusively, by recruitment of histone deacetylase activity. Thus, histone deacetylases appear to play an important gene-dependent regulatory role in the regulation of ventilation-induced gene transcription, with the caveat that histones are not the only substrates of histone deacetylase isoenzymes.
Acknowledgments
The authors gratefully acknowledge the excellent technical assistance of D. Karp and K. Viertmann (Research Center Borstel, Borstel, Germany).
- Received October 13, 2006.
- Accepted July 7, 2007.
- © ERS Journals Ltd
References










