





Abstract
Mutations in genes encoding members of the transforming growth factor (TGF)-β superfamily have been identified in idiopathic forms of pulmonary arterial hypertension (PAH). The current study examined whether perturbations to the TGF-β/Smad2,3 signalling axis occurred in a monocrotaline (MCT) rodent model of experimental PAH.
Expression of the TGF-β signalling machinery was assessed in the lungs and kidneys of MCT-treated rodents with severe PAH by semi-quantitative reverse-transcription (RT)-PCR, real-time RT-PCR and immunoblotting. TGF-β signalling was assessed in the lungs and in pulmonary artery smooth muscle cells (PASMC) from MCT-treated rodents by Smad2 phosphorylation, expression of the connective tissue growth factor gene, activation of the serpine promoter in a luciferase reporter system and by the induction of apoptosis.
The expression of type1 TGF-β receptor (TGFBR) activin-A receptor-like kinase1, TGFBR-2, TGFBR-3 (endoglin), Smad3 and Smad4; as well as TGF-β signalling and TGF-β-induced apoptosis, were dramatically reduced in the lungs and PASMC, but not the kidneys, of MCT-treated rodents that developed severe PAH.
The current data indicate that the transforming growth factor-β/Smad2,3 signalling axis is functionally impaired in monocrotaline-treated rodents with severe pulmonary arterial hypertension, underscoring the potential importance of transforming growth factor-β/Smad2,3 signalling in the onset or development of pulmonary arterial hypertension.
- Apoptosis
- monocrotaline
- pulmonary artery smooth muscle cells
- transforming growth factor-β
- vascular remodelling
Pulmonary vasoconstriction, vascular remodelling and thrombosis are the underlying causes of increased pulmonary vascular resistance, and thus the elevated pulmonary artery pressure observed in patients with pulmonary arterial hypertension (PAH) 1, 2. The vascular remodelling observed in small, normally nonmuscularised, pulmonary arteries involves an increase in pulmonary artery smooth muscle cell (PASMC) mass, secondary to endothelial dysfunction. Together, these changes result in medial hypertrophy, concentric obliteration of the lumen, and the formation of complex vascular structures known as plexiform lesions 3.
Although PAH can be inherited (familial; FPAH) or occur sporadically (idiopathic; IPAH) 4, the pathophysiological mechanisms at play during vascular remodelling in PAH have not been identified. Candidate effector molecules include serotonin, angiopoietin and also bone morphogenetic proteins (BMPs), which form a branch of the transforming growth factor (TGF)-β superfamily of growth factors 1, 2. Mutations in the gene encoding the type II BMP receptor are considered, at least in part, to be the genetic basis of FPAH, and BMP signalling has consequently received much attention in this context 1, 2, 5. In contrast, signalling by the TGF-β branch of this superfamily of growth factors has received comparatively little attention 6.
TGF-β signalling is initiated by binding of TGF-β ligand to the TGF-β receptor (TGFBR)-2 7. This promotes complex formation with one of two type I TGFBRs, either TGFBR-1 (activin-like kinase (ALK)-5) or activin-A receptor-like kinase (Acvrl1; ALK-1) 7. Complex formation may be assisted by accessory type III TGFBRs, either TGFBR-3 (betaglycan) or endoglin (the eng gene product, CD105). Signals are transmitted by intracellular signalling Smad molecules, notably Smads 2, 3 and 4 7 and TGF-β signalling may be antagonised by the inhibitory Smads, Smad6 and Smad7 7.
Mutations and polymorphisms in genes encoding TGF-β signalling machinery, and their functional consequences, have been implicated in many lung pathologies, including sarcoidosis 8 and chronic obstructive pulmonary disease (COPD) 9. With respect to PAH, mutations have been identified in eng 10 and acrvl1 11, 12 genes in patients with hereditary haemorrhagic telangiectasia and coexistent PAH, as well as in children with IPAH 13. Somatic mutations in TGFBR-2 have been reported in endothelial cells within plexiform lesions of IPAH patients 14. Therefore, mutations in all three types of TGF-β receptors have been described in PAH. Furthermore, reduced expression of genes encoding TGF-β3 and TGFBR-3 has been demonstrated in IPAH patients 15. Endothelial cells of plexiform lesions in IPAH patients exhibit reduced expression of TGF-β receptors and Smad proteins 6, and dysregulated TGF-β signalling is observed in PASMC, where TGF-β promoted the proliferation of PASMC from IPAH patients 16, but exhibited an opposite (growth inhibitory) effect on PASMC from healthy donors 16, 17. Together, these data overwhelmingly implicate dysregulated TGF-β signalling in PAH, although, to date, no study has examined TGF-β/Smad signalling in an animal model of PAH. In the current study, the authors set out to investigate whether perturbations to the TGF-β/Smad2 signalling axis occurred during monocrotaline (MCT)-induced pulmonary hypertension, a popular but poorly understood animal model for human PAH.
MATERIALS AND METHODS
Animals and haemodynamics
The animal ethics authority of the government of the State of Hessen (Germany) approved all animal procedures. Pulmonary hypertension was induced in adult male Sprague-Dawley rats by a single intraperitoneal injection of MCT (60 mg.kg−1; Sigma, St. Louis, MO, USA). Haemodynamic measurements and lung extraction were performed as described previously 18.
RNA isolation, semi-quantitative and real-time PCR
Total RNA was isolated from fresh lung tissue using a Qiagen RNeasy kit (Qiagen, Hilden, Germany), and screened by semi-quantitative reverse transcription (RT) PCR 19 using the intron-spanning primers indicated in table 1⇓. Cycle numbers lie in the logarithmic phase for each PCR. Quantitative changes in gene expression were also analysed by real-time RT-PCR by the ΔΔCt method 20, using the primer pairs indicated in table 1⇓ and the hydroxymethylbilane synthase (hmbs) gene as a reference.
Primers employed for reverse transcription PCR. Forward and reverse primers are indicated for real-time (rt) and semi-quantitative (sq) PCR reactions
Protein isolation and immunoblotting
Protein extraction, gel electrophoresis and immunoblotting were performed as described previously 19, 21. Immunoblots were probed with antibodies to Acvrl1 (1 µg.mL--1; R&D Systems, Minneapolis, MN, USA), TGFBR-1 (1:1500) and TGFBR-2 (both 1:1000; Santa Cruz, San Francisco, CA, USA), phospho-Smad2 (Ser465/467), Smad3 and Smad4 (all 1:1000; Upstate, Charlottesville, VA, USA), Smad2 (1:1000; Zymed, San Fransisco, CA, USA), and β-actin (1:1000; Cell Signaling Technology, Danvers, MA, USA). Peroxidase-conjugated secondary antibodies were from R&D Systems (Wiesbaden, Germany).
Immunohistochemistry
Elastin and haematoxylin and eosin staining, and immunofluorescence were performed on 3 μm tissue sections 18, with antibodies against smooth muscle actin (1:850; Sigma, Taufkirchen, Germany), von Willebrand factor (vwf; 1:800; Dako, Hamburg, Germany), TGFBR-1 (R-20), TGFBR-2 (H-567) and proliferating cell nuclear antigen (PCNA; all at 1:100; Santa Cruz), Smad3 and Smad4 (both at 1:50; Upstate), Smad2 (R&D Systems) and phospho-Smad2 (Cell Signaling Technology). Immune-complexes were visualised with a Histostain Plus Kit (Zymed). Immunohistochmemistry was quantified by counting the number of positive cells as a percentage of the total number of cells counted. It is important to stress that these data reflect absolute changes (positive versus negative staining) in a signal and do not reflect intermediate changes (strong versus weak staining) in signal intensity; therefore, data must be interpreted with caution.
Luciferase reporter assay
Primary rat PASMC were isolated from the second to the fifth branch of the pulmonary artery as described previously 18, from saline-treated control or MCT-treated rats. Early passage (first two passages) PASMC, seeded in 24-well plates (70% confluent), were transiently transfected with LipofectAMINE (Invitrogen, Karlsruhe, Germany) as described for human PASMC 22, with p(CAGA)12-luc, a reporter construct that contains a TGF-β-responsive promoter derived from the serpine1 gene, located upstream of a firefly luciferase gene 23. Alternatively, cells were transfected with paraganglioma (pGL3)-basic (containing a promoterless luciferase gene) or pGL3-control (containing a constitutively-expressed luciferase gene), as negative and positive controls, respectively (both from Promega, Mannheim, Germany). Cells were incubated with 2 ng·mL−1 TGF-β1 (R&D Systems) for 12 h before the firefly luciferase activity was assessed 19. Values were normalised for the transcriptional activity of the pGL3-control vector.
Assessment of apoptosis of PASMC
Early passage PASMC were seeded at 1×104 cells·well−1 in chamber slides and grown to ∼80% confluence. Quiescent cells (cultured in 0.1 % (volume/volume) foetal bovine serum for 48 h) were treated with TGF-β1 (2 ng·mL−1, 24 h) and apoptosis was assessed using a terminal deoxyribonucleotidyl transferase-mediated deoxyuridine triphosphate nick end-labelling (TUNEL) assay (Roche, Indianapolis, IN, USA) 18. Cells were scored for TUNEL and 4′,6′-diamino-2-phenylindole staining in 20 random fields per sample.
Statistical treatment of data
Data are presented as mean±sd. Differences between groups were analysed by ANOVA and the Student–Newman–Keul post hoc test for multiple comparisons, with a p-value <0.05 regarded as significant.
RESULTS
MCT causes severe PAH and vascular remodelling in rats
Administration of MCT caused pronounced vascular remodelling and PAH in rats 4 weeks after administration. Vascular remodelling was evident by thickening of the vessel walls of the small pulmonary arteries (fig. 1a–d⇓), together with a significant (44%) increase in the ratio of right ventricle-to-left ventricle and septum (table 2⇓), indicative of right heart hypertrophy. These changes were accompanied by a two-fold increase in right ventricular systolic pressure (table 2⇓) indicative of severe PAH, and a 30% decrease in the arterial oxygen tension (table 2⇓) although no significant change (11% reduction) in systemic arterial pressure was observed (table 2⇓).

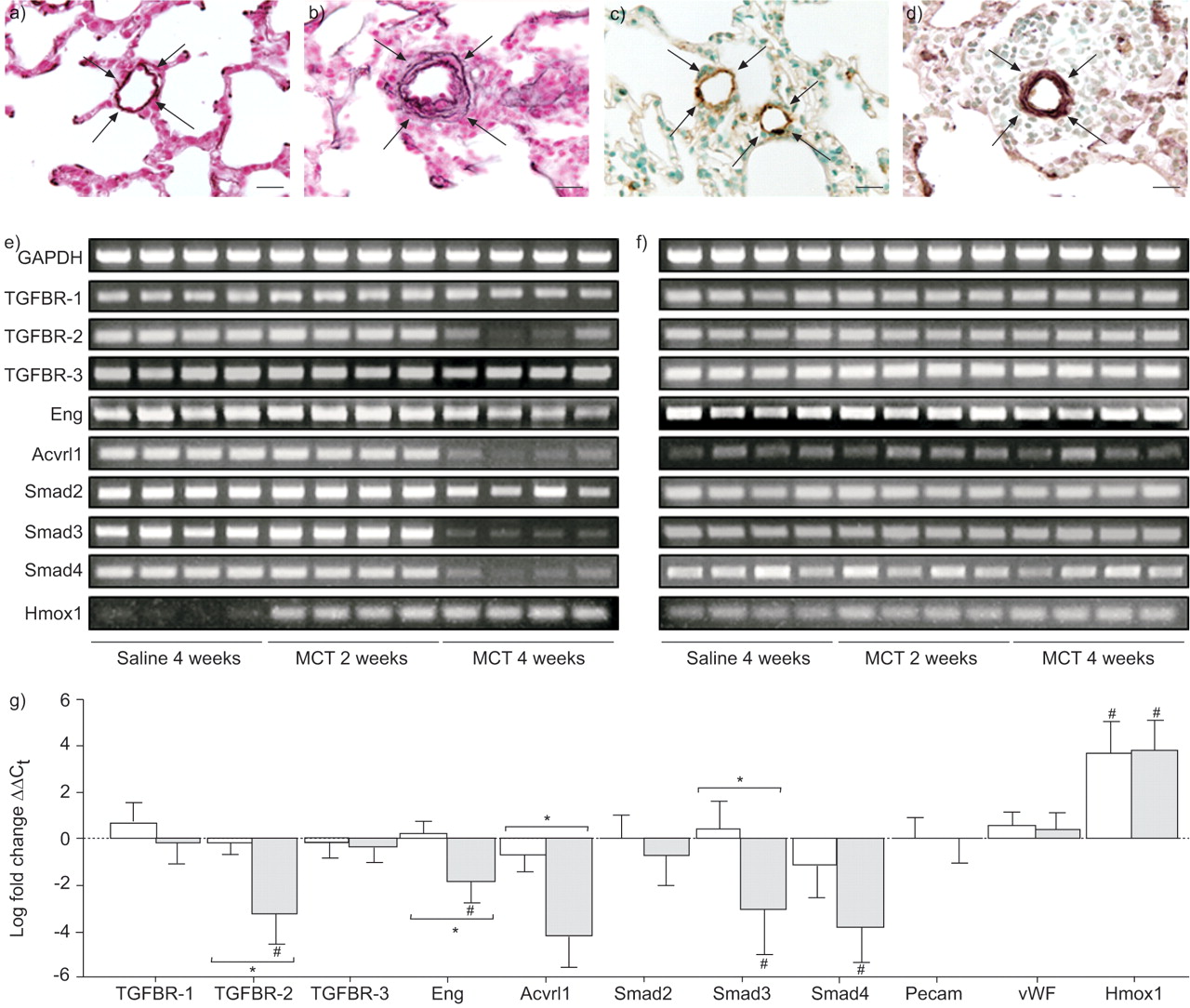
Monocrotaline (MCT)-induced pulmonary vascular remodelling and changes in expression of the transforming growth factor (TGF)-β signalling machinery. Changes in medial wall thickness and vessel muscularisation were investigated in small pulmonary arteries in rats 28 days after saline (a, c) or MCT (b, d) administration. Changes in wall thickness (a, b) were evident by elastin staining (arrows), and vessel muscularisation (c, d) was visualised by dual staining for von Willebrand factor (brown) and smooth muscle actin (purple), with a haematoxylin counterstain. Scale bars = 20 μm. Changes in the expression of genes encoding the TGF-β signalling machinery were assessed in the e) lungs and f) kidneys of saline- or MCT-treated rats by semi-quantitative reverse transcription (RT)-PCR and g) by quantitative real-time RT-PCR, 2 weeks (□) and 4 weeks (░), relative to saline-treated controls at 4 weeks. Data represent the mean±sd. TGFBR: TGF-β receptor; Acvrl1: activin-A receptor-like kinase; Pecam: platelet-endothelial cell adhesion molecule; vWF: von willebrand factor; Hmox: haem oxygenase. #: p<0.05 relative to controls; *: p<0.05 between indicated groups (n = 4, per group).
Induction of pulmonary hypertension in rats by intraperitoneal monocrotaline(MCT)
Expression of TGF-β signalling machinery is reduced in the lungs of MCT-treated rats
A dramatic decrease in expression of mRNA encoding TGFBR-2, endoglin, Acvrl1, Smad3 and Smad4 was observed by real-time RT-PCR in the lungs of rats 4 weeks after MCT treatment (fig. 1g⇑). Similarly, downregulated expression of genes encoding TGFBR-2, Avcrl1, Smad3 and Smad4 was observed by semi-quantitative RT-PCR (figs 1e⇑ and f) in the lungs, but not kidneys, of rats 4 weeks after MCT administration. The mRNA levels of the endothelial markers platelet–endothelial cell adhesion molecule 1 and vwf were unaltered at 2 and 4 weeks post-MCT treatment (fig. 1g⇑), indicating no major loss of endothelium at these time-points. The haem oxygenase 1 gene was employed as a control, since its expression is elevated in the lungs of MCT-treated rats that develop PAH 24.
Since changes in mRNA expression do not always reflect changes in the protein expression 25, protein expression was also assessed by immunoblotting. Consistent with the RT-PCR data, significantly reduced levels of TGFBR-2, Avcrl1, Smad3 and Smad4 were observed in the lungs of MCT-treated rats 4 weeks after MCT administration (fig. 2⇓).

a) Changes in the protein expression of components of the transforming growth factor (TGF)-β signalling machinery in the lungs of monocrotaline (MCT)-treated rats were assessed by immunoblot. b–g) Quantitation of immunoblotting data for b) TGF-β receptor (TGFBR)-1, c) TGFBR-2, d) activin-A receptor-like kinase 1(Acvrl1), e) Smad2, f) Smad3 and g) Smad4. C:control. *: p<0.05 relative to controls (n = 4, per group).
Antibodies against Acvrl1 did not yield any signal over background staining on lung sections (data not shown). However, both TGFBR-1 and TGFBR-2 exhibited strong staining in the airway epithelium in saline-treated rats (figs 3a–d⇓). Staining for TGFBR-1 was also evident in the smooth muscle and endothelium of saline-treated rats (figs 4c⇓ and d). In the case of TGFBR-1, the staining exhibited similar intensity and pattern in MCT-treated rats, although the background was higher due to intense inflammtory cell infiltration (figs 3a⇓, 4a and b). However, staining for TGFBR-2 was noticeably weaker in the airway epithelial layer (fig. 3d⇓), and tended towards reduced levels in the vascular smooth muscle layer of small vessels of MCT-treated rats (fig. 5b⇓). These data are consistent with the current observations that mRNA and protein expression of TGFBR-1 and TGFBR-2 are reduced in the lungs of MCT-treated rats.
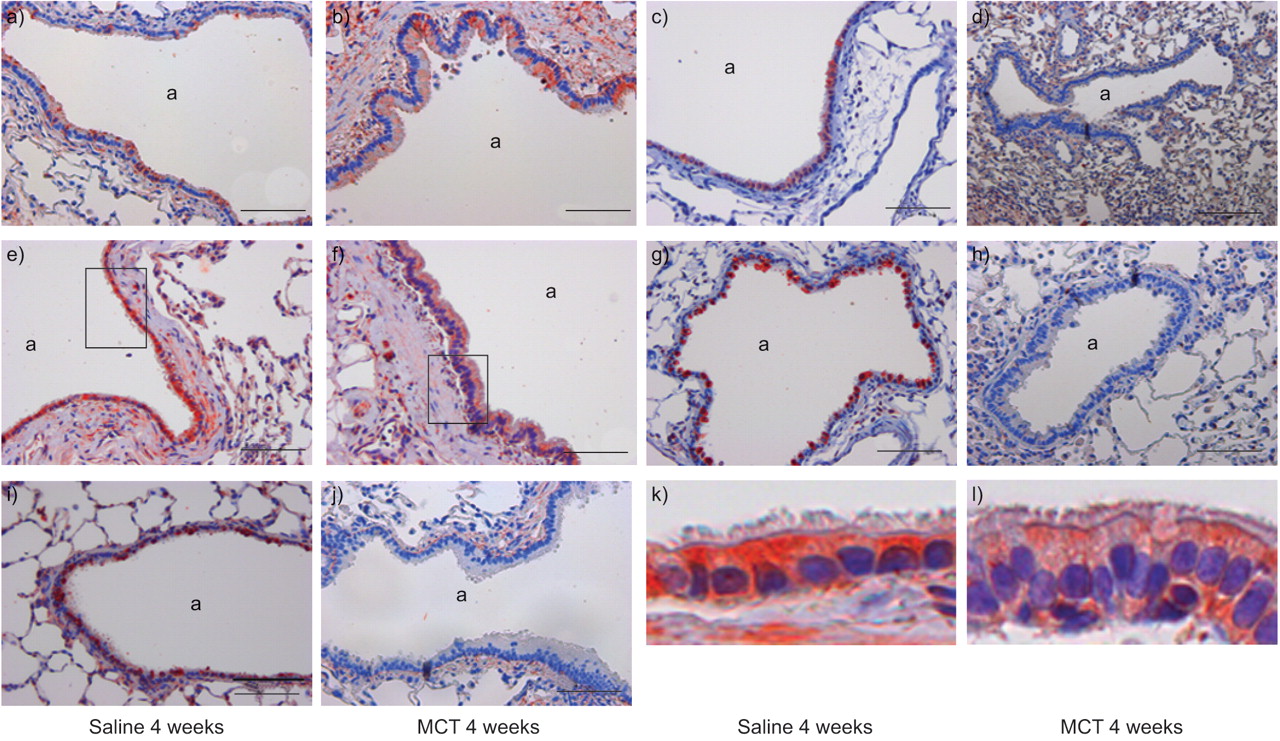
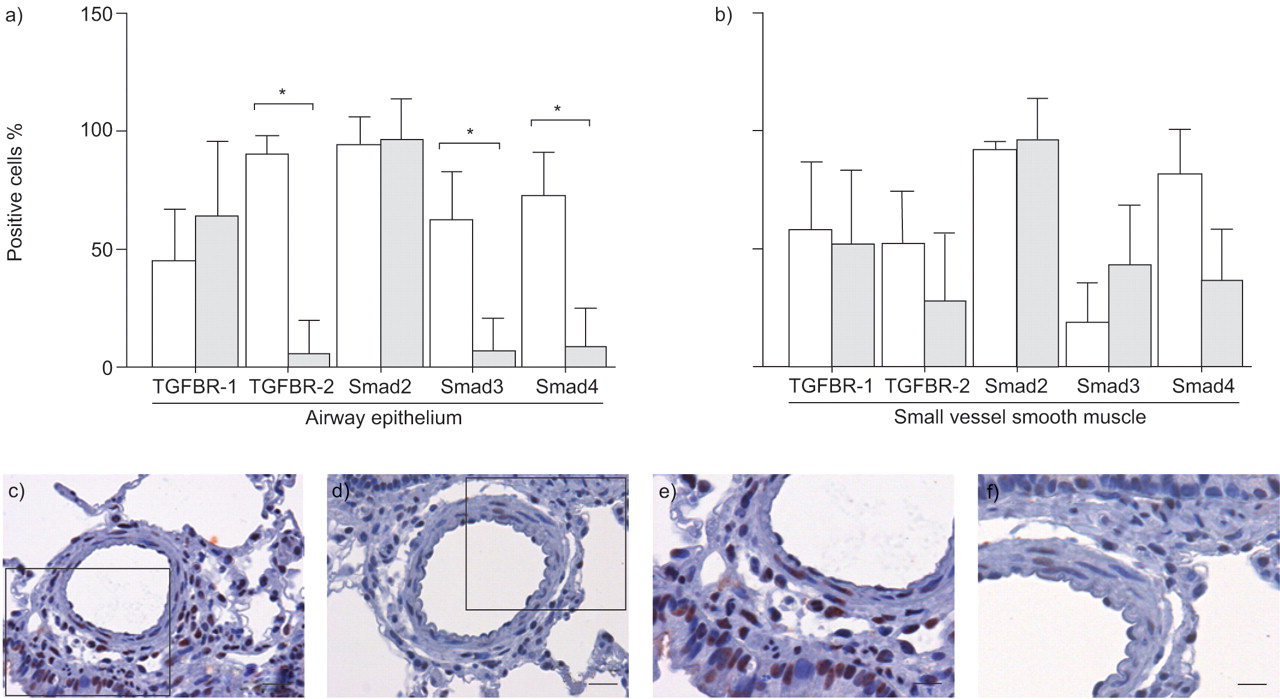
Localisation of: a, b) transforming growth factor-β receptor (TFGBR)-1; c, d) TGFBR-2; e, f) Smad2; g, h) Smad3; and i, j) Smad4 in the airways of lungs from saline- and monocrotaline (MCT)-treated rats, 4 weeks after treatment. k and l) enlarged boxed sections of the epithelium from e) and f), respectively. a: Airway. Scale bars = 100 μm.

Localisation of: a–d) transforming growth factor-β receptor (TGFBR)-1; e–h) TGFBR-2; i–l) Smad4; m–o) Smad3; and p–r) Smad2 in pulmonary resistance arteries of rats, 4 weeks after treatment with saline or monocrotaline (MCT). Large diameter vessels had an internal diameter 400–800 µm. a, c, e, g, I, k, m–r) Scale bar = 100 μm. Small diameter vessels were those with an internal diameter between 150 and 400 μm. b, d, f, h, j, l) Scale bar = 50 μm. v: Blood vessels; a: Airways.

Assessment of transforming growth factor (TGF)-β signalling in the lungs of saline- and monocrotaline (MCT)-treated rats. The expression of TGF-β and Smad proteins was quantitated in a) the airway epithelium and b) smooth muscle layer of small vessels in the lungs of saline-treated (□) and MCT-treated (░) rats, 4 weeks after treatment. Data are presented as the mean±sd of positive cells in eight representative fields. *: p<0.05, between indicated groups. Localisation of phospho-Smad2 in the lungs of c, e) saline- and d, f) MCT-treated rats. Arrowheads indicate positive staining. TGFBR: TGF-β receptor. c, d) Scale bars = 50 μm. e, f) Scale bars = 25 μm.
In the case of Smad proteins, staining for Smad2 was strong, particularly in the airway epithelium of both saline- and MCT-treated rats (figs 3e⇑ and f, and 5a). In contrast, while intense staining for Smad3 and Smad4 in the airway epithelium was observed for saline-treated rats (figs 3g⇑ and i, and 5a⇑), this staining was markedly reduced in MCT-treated rats (figs 4h⇑ and j, and 5a⇑). Of interest, Smad4 exhibited a strong nuclear staining in the endothelium of saline-treated rats (fig. 4l⇑), in ∼77±6 % of cells counted, while this staining was largely lost in MCT-treated rats (8±4 % of cells counted). Similarly, Smad4 staining exhibited a reduced trend in the vascular smooth muscle of MCT-treated rats (fig. 5b⇑); however, this trend did not approach statistical significance. These data support the observation that the mRNA and protein levels of Smad3 and Smad4 are reduced in MCT-treated rats.
The TGF-β/Smad2,3 signalling axis is impaired in the lungs of MCT-treated rats
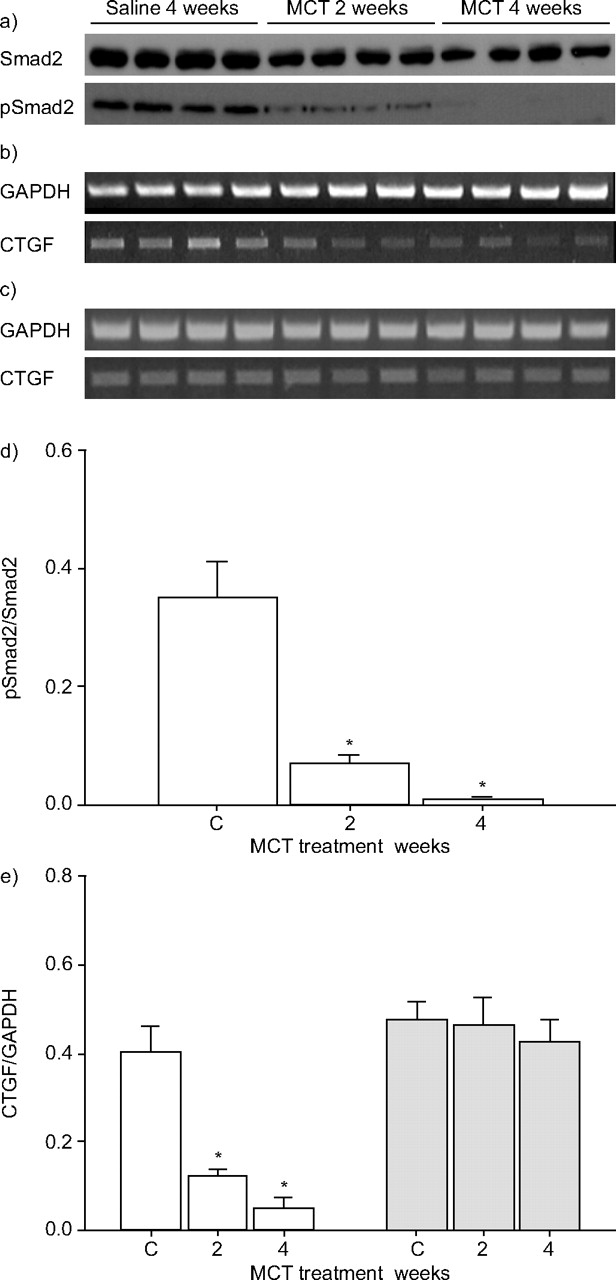
The current authors’ observation that MCT administration caused a dramatic reduction in the expression of TGF-β receptors and Smads, suggested that TGF-β signalling would be perturbed in the lungs of MCT-treated rats. Indeed, while phospho-Smad2 staining was evident in the vascular smooth muscle and airway epithelium of lungs from saline-treated rats, this staining was largely lost in lungs from MCT-treated rats (fig. 5c–f⇑). Furthermore, baseline levels of phospho-Smad2 (although not total Smad2 levels) were significantly reduced in whole lung extracts from MCT-treated rats (fig. 6a⇓ and d) suggesting functional impairment of the TGF-β/Smad2,3 signalling axis. Interestingly, this decrease was evident before changes in TGFBR or Smad expression were evident by immunoblot (fig. 2a⇑), suggesting that other mechanisms are at play, which downregulate TGF-β/Smad2 signalling (perhaps in response to the massive inflammation in lungs at 2 weeks post-MCT adminsitration), or that the receptor and Smad expression changes were too subtle to be evident by immunoblot at this time-point.
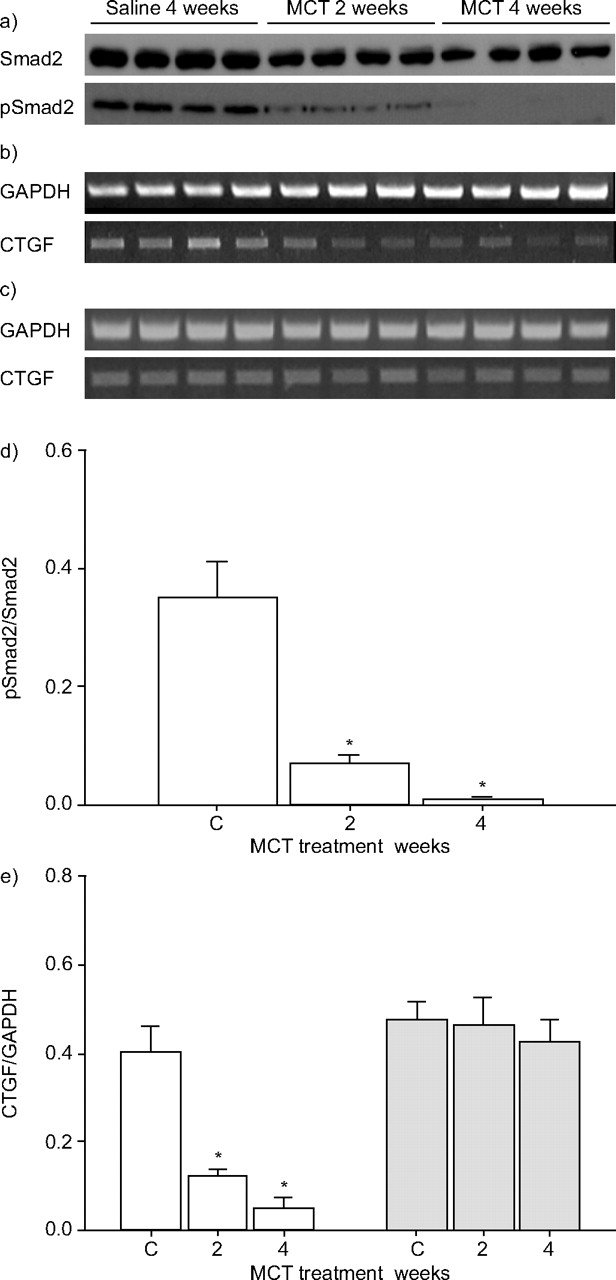
Transforming growth factor (TGF)-β signalling assessed in the lungs of saline- and monocrotaline (MCT)-treated rats by a) phospho-Smad2 phosphorylation and b, c) expression of the connective tissue growth factor (CTGF) and the housekeeping reduced glyceraldehyde phosphate dehydrogensae (GAPDH) genes in lung (b) and kidney (c). These data were quantified by densitometry in d) and e), respectively. □: lung; ░: kidney. *: p<0.05 relative to controls (C; n = 3–4 per group).
Together, these data suggest that the expression of TGF-β-induced genes in the lung would also be downregulated. To test this idea, the authors examined mRNA levels of the connective tissue growth factor (CTGF) gene, which is regulated by TGF-β. As expected, ctgf mRNA levels were significantly lower in the lungs of MCT-treated rats, although not in the kidneys (figs 6b⇑, c and e). Therefore, these data indicate that MCT administration specifically perturbs TGF-β signalling in the lungs of rats in the current experimental model of PAH.
Given the important role of TGF-β in growth-regulation, cell proliferation was also assessed in lungs from MCT-treated rats by staining for the PCNA. Few proliferating cells were evident in lungs of saline-treated rats (figs 7a⇓ and d). However, 2 weeks after MCT administration, increased cell proliferation was observed in the alveolar septae, smooth muscle and endothelium of the lung (figs 7b⇓ and e). Additionally, massive inflammatory cell infiltration was evident 2-weeks post-MCT administration, evident from a dramatically increased number of macrophages (fig. 7b⇓). Four weeks after MCT administration, proliferating cells were primarily detected in the smooth muscle layer of pulmonary arteries, although proliferating cells were also evident in the airway epithelium and the interstitium (figs 7c⇓ and f).
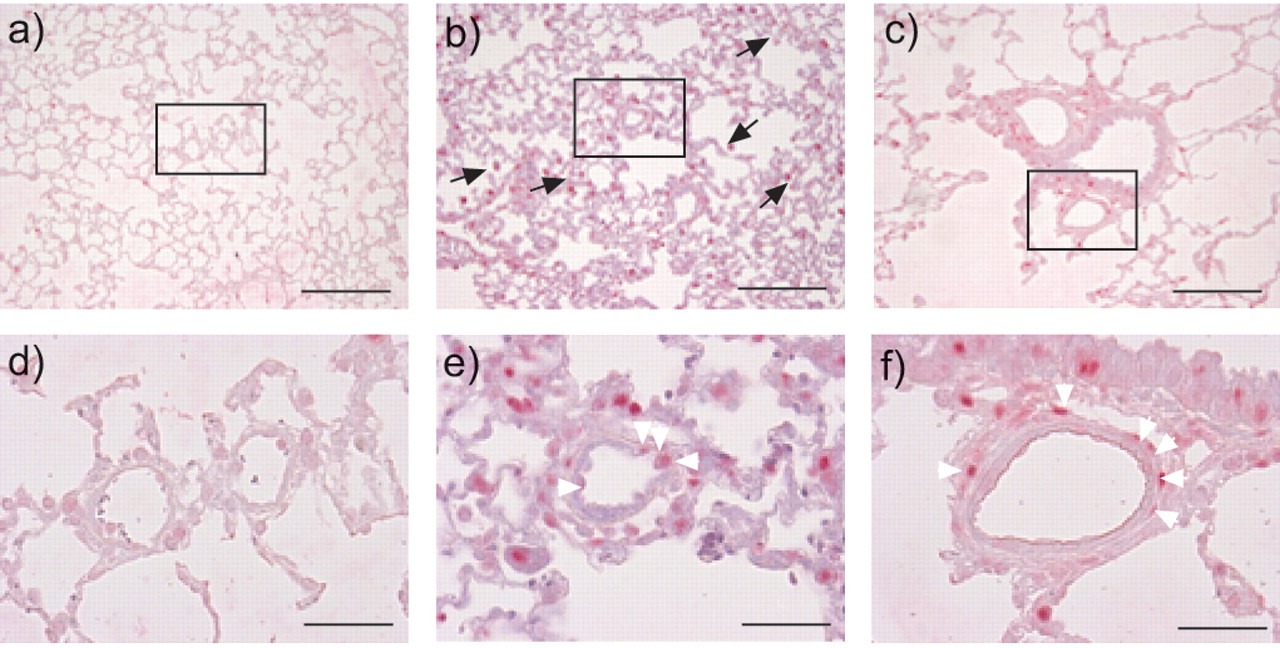
Assessment of cell proliferation in the lungs of a, d) saline- and b, c, e, f) monocrotaline (MCT)-treated rats by proliferating-cell nuclear antigen (PCNA) staining, 2 weeks (b, e) and 4 weeks (c, f) after MCT treatment. d–f) Enlarged boxed sections in a–c), respectively. White arrowheads indicate positive PCNA staining in the smooth muscle layer of small resistance arteries; black arrows indicate macrophages. a–c) Scale bars = 200 μm. d–f) Scale bars = 50 μm.
The TGF-β/Smad2,3 signalling axis is impaired in PASMC from MCT-treated rats
The current data have so far relied on experiments in whole lung homogenates, which reflect the net TGF-β signalling capacity of the endothelium, epithelium, smooth muscle, fibroblast and inflammatory cell composition of the lung. The current authors felt it relevant to examine TGF-β signalling in a single cell type. Since PASMC, which are responsive to TGF-β 16, 17, are accredited with a key role in the pathogenesis of familial and nonfamilial forms of PAH 1, and increased PASMC proliferation is observed in the lungs of rats after MCT treatment, TGF-β signalling was examined in PASMC isolated from the lungs of saline- or MCT-treated rats (fig. 8⇓).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
a) Transforming growth factor (TGF)-β signalling was impaired in pulmonary artery smooth muscle cells (PASMC) from monocrotaline (MCT)-treated rats, evident from reduced baseline phospho-Smad2 phosphorylation in nonquiescent cells. b) Reduced activity of the TGF-β-responsive element of the serpine1 promoter in response to TGF-β1 stimulation. c) Reduction in the number of apoptotic nuclei observed in PASMC from MCT-treated (░) versus saline-treated (□) rats after TGF-β1 stimulation. *: p<0.05 between indicated groups.
Baseline phospho-Smad2 levels (but not total Smad2 levels) were reduced in PASMC from MCT-treated rats, indicating that the TGF-β/Smad2 axis was also functionally impaired in PASMC. Specific induction of TGF-β signalling was investigated by transfecting the p(CAGA)12-luc reporter plasmid 23 into PASMC derived from saline- or MCT-treated rats. This plasmid contains a firefly luciferase gene, located downstream of a TGF-β-responsive promoter derived from the serpine1 gene. Therefore, the induction of luciferase activity by TGF-β is a measure of TGF-β signalling. The ability of TGF-β to induce luciferase expression was markedly (two-fold) lower in PASMC from MCT-treated rats (fig. 8b⇑), confirming that the TGF-β/Smad axis is functionally impaired in PASMC from MCT-treated rats.
Since TGF-β can induce apoptosis in PASMC 17, it was reasoned that this effect would be less apparent in PASMC from MCT-treated rats. Indeed, a more than two-fold reduction in the induction of apoptosis by TGF-β was observed in PASMC from MCT-treated rats, as assessed by TUNEL staining (fig. 8c⇑). These data indicate that functional impairment of the TGF-β/Smad signalling axis in PASMC from MCT-treated rats is physiologically relevant. This idea is supported by the increased PASMC proliferation in the lungs of MCT-treated rats reported in the current study (fig. 7⇑) and the complete absence of apoptosis in PASMC noted in MCT-treated rats in another study 18.
DISCUSSION
A growing body of evidence points to a key role for dysregulated signalling by the BMP/Smad1,5,8 axis of the TGF-β superfamily in the onset or development of PAH 1, 2, 4, 6, 12–16. In contrast, a role for the TGF-β/Smad2,3 axis in PAH has been relatively unexplored. Therefore, the aim of the current study was to establish whether the TGF-β/Smad signalling axis was impaired in MCT-induced PAH in rats, a popularly used, albeit poorly understood, model for familial and idiopathic PAH.
The current study documents a dramatic downregulation of expression of both TGFBRs, and some of their associated Smad proteins, 4 weeks after MCT administration, by which time pronounced pulmonary vascular remodelling and PAH were evident. Amongst the receptors, a dramatic decrease in Acvrl1 and TGFBR-2 expression was evident, while expression of TGFBR-1 was unchanged. Similarly, expression of two key TGF-β intracellular signalling molecules, Smad3 and Smad4, was decreased. These changes were restricted to the lung, since they were not observed in the kidneys of MCT-treated rats. Furthermore, levels of both phosphorylated Smad2 and mRNA encoding CTGF were also downregulated, indicating diminished TGF-β signalling in the lungs, but not the kidneys, of MCT-treated rats with severe PAH. In PASMC isolated from MCT-treated rats, both baseline and induced TGF-β signalling were also reduced, compared with PASMC from saline-treated controls. This impaired TGF-β signalling was physiologically relevant, since PASMC from MCT-treated rats were resistant to the pro-apoptotic effects of TGF-β. Taken together, the current data demonstrate dramatically impaired signalling by the TGF-β/Smad axis in the lungs of MCT-treated rats with severe PAH.
Four-fold elevated levels of TGF-β have been described during MCT-induced PAH in rats 26, and it has been suggested that this may underlie the increased expression of some extracellular matrix genes in this model. The present data would argue that this is not the case, but rather that the two-fold increase in mRNA encoding tropoelastin, fibronectin and collagen Iα1 levels observed in that previous study 26 may have been induced by other fibrogenic cytokines, possibly interleukin-1α, 1β and tumour necrosis factor-α, since levels of all three cytokines are elevated in MCT-induced PAH. Interestingly, induction of a dominant-negative TGFBR-2 gene in hypoxia-induced PAH attenuated hypoxia-driven vascular remodelling and pulmonary hypertension 27, which suggests a protective role for TGFBR-2 in hypoxia-driven PAH, in contrast to the current data, which correlates reduced TGFBR-2 with increased abberant vascular remodelling in MCT-induced PAH. Together, these data emphasise that the vascular remodelling in these two models is very different 28, an idea underscored by the recent observation that bone marrow-derived progenitor cells can limit pulmonary vascular remodelling induced by MCT, but not that induced by hypoxia 29.
A common feature of the vascular remodelling observed in all forms of PAH is the increased muscularisation of small pulmonary arteries 1, 2. This has, in part, been attributed to the aberrant growth of PASMC, which exhibit impaired responses to stimuli that control proliferation and apoptosis of PASMC. TGF-β is one of those stimuli, promoting the apoptosis of healthy PASMC 17 and inhibiting the proliferation of both healthy PASMC and PASMC from patients with Eisenmenger's syndrome who exhibit PAH secondary to ventricular septal defects 16. However, PASMC from patients with IPAH or FPAH are resistant to the anti-proliferative effects of TGF-β 16, suggesting that the failure of TGF-β to control PASMC growth in PAH may, in part, underlie the increased muscularisation of normally nonmuscularised small pulmonary arteries of patients with FPAH or IPAH. Data presented in the present study support this idea, since increased muscularisation of small pulmonary arteries was also observed in MCT-treated rats with severe PAH, and PASMC from these rats were insensitive to the pro-apoptotic effects of TGF-β. Thus, impaired TGF-β signalling in PASMC appears to be a common feature of human IPAH and MCT-induced PAH.
In addition to PASMC, dysregulated TGF-β signalling is also likely to affect the endothelium. Endothelial cells are peculiar in that they express two different type I TGFBRs: TGFBR-1 and Acvrl1. These two receptors mediate the opposing effects of TGF-β on endothelial function, and together they balance the activation state of the endothelium. Signalling by Acvrl1 promotes endothelial cell proliferation and migration 30, and endoglin plays a key role in promoting these effects 31, thereby stimulating angiogenesis. In contrast, signalling by TGFBR-1 opposes Acvrl1/endoglin signalling, inhibits endothelial cell proliferation and migration and maintains the endothelium in a quiescent state 30. The current data demonstrated a significant downregulation of both Acvrl1 and endoglin expression in the lungs of MCT-treated rats, while TGFBR-1 expression remained unchanged. These data suggest that, in the context of TGF-β signalling, the balance is tipped strongly in favour of TGFBR-1 signalling, which would maintain a quiescent endothelium in MCT-induced PAH. This idea is supported by the observation that MCT has a cytostatic effect on the rat pulmonary endothelium 32. Clearly, however, this idea remains speculative since isolated endothelial cells were not examined in the present study.
The data presented here support a role for reduced TGF-β signalling in promoting PAH. This idea gains credence from observations that the converse situation, i.e., increased TGF-β signalling, can afford protection against PAH. For example, in patients with very severe (Global Initiative for Chronic Obstructive Lung Disease stage IV) COPD, the expression of Smads 2,3 and 4 was normal, while that of the inhibitory Smad, Smad7, was reduced 33, which would lead to potentiated TGF-β signalling. In a separate study it was demonstrated that expression of TGFBR-2 was upregulated in the pulmonary arteries of patients with very severe COPD 34. Together, these studies suggested that TGF-β signalling is increased in COPD, leading Beghe et al. 34 to propose that upregulation of TGF-β signalling in severe COPD has a protective (anti-proliferative) role, perhaps explaining the low incidence of severe pulmonary hypertension seen in COPD patients 35.
SUMMARY
The current data suggest that dramatically reduced expression of components of the transforming growth factor-β signalling machinery results in impaired transforming growth factor-β signalling in the lungs of monocrotaline-treated rats. This dysregulated transforming growth factor-β signalling may underlie the aberrant proliferation of pulmonary artery smooth muscle cells seen in monocrotaline-induced pulmonary arterial hypertension; and also has implications for the activation state of the endothelium. The current data strongly support a role for impaired transforming growth factor-β signalling in the onset and developments of sporadic and familial forms of idiopathic pulmonary arterial hypertension, in which germline mutations in genes encoding the transforming growth factor-β signalling machinery have already been reported.
Statement of interest
The authors are supported by the Deutsche Forschungsgemeinschaft (SFB547 Cardiopulmonary Circulation), the Excellence Cluster Cardiopulmonary System of the Universities of Giessen and Frankfurt and the Max Planck Institute for Heart and Lung Research (Bad Nauheim, all Germany), and the European Commission Sixth European Framework Programme “Pulmotension” (University of Giessen, Germany).
- Received October 24, 2006.
- Accepted March 14, 2007.
- © ERS Journals Ltd
References










