





Abstract
High levels of particulate matter in ambient air are associated with increased respiratory and cardiovascular health problems. It has been hypothesised that it is the ultrafine particle fraction (diameter <100 nm) that is largely responsible for these effects. To evaluate the associated mechanisms on a molecular level, the current authors applied an expression profiling approach.
Healthy mice were exposed to either ultrafine carbon particles (UFCPs; mass concentration 380 µg·m-3) or filtered air for 4 and 24 h. Histology of the lungs did not indicate any pathomorphological changes after inhalation.
Examination of the bronchoalveolar lavage fluid revealed a small increase in polymorphonuclear cell number (ranging 0.6–1%) after UFCP inhalation, compared with clean air controls, suggesting a minor inflammatory response. However, DNA microarray profile analysis revealed a clearly biphasic response to particle exposure. After 4 h of inhalation, mainly heat shock proteins were induced, whereas after 24 h, different immunomodulatory proteins (osteopontin, galectin-3 and lipocalin-2) were upregulated in alveolar macrophages and septal cells.
In conclusion, these data indicate that inhalation of ultrafine carbon particles triggers a biphasic pro-inflammatory process in the lung, involving the activation of macrophages and the upregulation of immunomodulatory proteins.
Epidemiological studies have shown that increased levels of particulate matter (PM) in ambient air are associated with aggravation of respiratory diseases and cardiovascular complications. A strong association has been seen for respiratory and cardiac deaths, particularly among elderly people 1–3. Oxidative stress, induced by inhaled particles, successively leading to activation of pro-inflammatory gene transcription, is one mechanism thought to cause the adverse health effects of ambient PM. In particular, alveolar macrophages may be activated by particles and release cytokines and reactive oxygen species 4.
Different hypotheses were developed to explain which particle properties drive the adverse health effects. These hypotheses deal with the particle’s charge, its content of transition metals, and its size and specific surface area. According to the “ultrafine hypothesis of particle toxicity”, ambient ultrafine particles (UFP), i.e. particles with a diameter <100 nm, are the proportion of particulate air pollution that mainly causes the adverse health effects 5. Animal studies indicate that at high exposure levels ultrafine carbon particles (UFCPs) and titanium dioxide particles have a greater toxic potential than fine particles with a diameter ranging between 0.1–1.0 µm 6, 7. UFP induce pulmonary inflammation at a lower mass concentration than larger particles 8. Although UFP comprise only 1–8% of the mass, they present up to 99% of the number of ambient PM 9. Due to their small size, UFP are known to enter the alveolar–capillary barrier 10, 11, translocate from the lung into the blood 12, and thus have the capability to directly interact with extrapulmonary organs.
So far, the molecular and cellular events induced by inhalation of UFP are poorly understood. The objective of the current study was to identify genes regulated by the exposure to UFP in order to elucidate pathways that are activated by these particles and whose activation possibly leads to adverse health effects.
Young, healthy BALB/cJ mice that were exposed to UFCPs were used as a model. The present authors performed DNA microarray analysis of whole-lung RNA from mice that were exposed to high levels of UFP for different periods of time. Among the affected genes, concerted upregulation of genes encoding for molecules involved in oxidative stress and macrophage activation were found. The current results are the first to demonstrate that inhalation of UFCPs leads to macrophage activation and triggers pro-inflammatory processes in healthy mice. Furthermore, the current authors identified the regulation of soluble immunomodulatory proteins that might serve as a useful marker for inflammatory processes induced by particle inhalation in further studies.
MATERIALS AND METHODS
Animals
Female BALB/cJ mice, established at The Jackson Laboratory (Bar Harbor, ME, USA), were shipped to the GSF-National Research Center for Environment and Health (Neuherberg, Germany) at 8 weeks of age. The animals were kept at the GSF in “isolated ventilated cages” (IVC-Racks; BioZone, Ramsgate, UK) supplied with filtered air in a 12-h light/12-h dark cycle. Specified pathogen-free status was approved by a health certificate according to Federation of European Laboratory Animals Science Association guidelines. Food and water were available ad libitum. Animals were aged 10–12 weeks when studied. Eight animals per experimental group were analysed. Experimental protocols were in accordance with the German Law on Animal Protection and approved by the Bavarian Animal Research Authority (approval no. 211-2531-108/99).
Particle generation and whole-body exposure chamber
The set-up of the whole-body exposure system for rodents has been described previously by Karg et al. 13. Briefly, the exposure chamber was supplied with a constant flow of humidified air (23°C, 46% relative humidity) and loaded with UFCPs. UFCPs were produced by an improved electric spark generator (Model GFG 1000; Palas, Karlsruhe, Germany) operated with ultrapure graphite electrodes in an argon atmosphere (<10-6 impurities) 14. UFCPs produced by this method consist of ≥96% elemental carbon 15. Particle number concentration (CPC 3022A; TSI, St. Paul, MN, USA) and size distribution (EMS 150; Hauke, Gmunden, Austria) were continuously monitored at the entrance of the exposure chamber. Particle number concentration was 8×106 cm-3 with a count median diameter of 49 nm. Mass concentration was measured gravimetrically by filter sampling. The average UFCP mass concentration in the exposure chamber was 380 µg·m-3.
Experimental groups
Groups of eight mice were exposed to UFCPs or clean air for 4 or 24 h, respectively. After particle or clean air exposure, bronchoalveolar lavage (BAL) was performed on eight mice per condition, and blood was taken from the same animals. Lungs were taken for RNA preparation from an additional eight animals per experimental condition. Eight further mice were sacrificed either after exposure to UFCPs or clean air for 24 h and different organs were taken for histological examinations.
BAL
After exposure, mice were anaesthetised by i.p. injection of a mixture of xylazine and ketamine, and killed by exsanguination. BAL was performed by cannulating the trachea and infusing the lungs 10 times with 1.0 mL of PBS without Ca2+ and Mg2+. The BAL fluid (BALF) from lavage one and two, as well as those from three to 10 were pooled and centrifuged (425×g for 20 min at room temperature). The cell-free supernatant from lavage one and two was used for the biochemical measurements. For each animal, the 10 cell pellets were unified and resuspended in 1 mL of RPMI 1640 Medium (BioChrome, Berlin, Germany) supplemented with 10% foetal calf serum (Seromed, Berlin, Germany), and the number of living cells was determined by the trypan blue exclusion method. The cell differentials were performed on cytocentrifuge preparations (May–Grünwald–Giemsa staining, 2×200 cells counted). Polymorphonuclear leukocytes were used as inflammatory markers.
Biochemical analyses
Lactate dehydrogenase (LDH) activity was assayed spectrophotometrically by monitoring the reduction of oxidised nicotinamide adenine dinucleotide at 366 nm in the presence of lactate. Total proteins were determined spectrophotometrically at 620 nm applying the BioRad Protein Assay Dye Reagent (Nr.500-0006; BioRad, Munich, Germany).
RNA purification
Frozen lungs were thawed in lysis buffer (supplied with RNA isolation kit) and homogenised with a FastPrep FP120 cell disrupter (BIO101/Savant; Qbiogene, Heidelberg, Germany) for 40 s. The RNA was isolated by using the RNeasy kit (Qiagen, Hilden, Germany). A DNase I treatment was routinely performed. For each experimental condition, RNA from the lungs of eight animals was prepared.
Microarray analysis
The GeneChip hybridisations were carried out by a service provider (RZPD; Deutsches Ressourcenzentrum für Genomforschung GmbH, Berlin, Germany). The RNA was quantified and equal amounts of RNA from eight lungs per experimental condition were pooled. For sample preparation, 15 µg of total RNA were used. First-strand synthesis was carried out by a T7-(dT)24 primer and Superscript II reverse transcriptase (Invitrogen Life Technologies, Karlsruce, Germany). Second-strand synthesis was performed according to the Superscript Choice system. Biotin-labelled cRNA was generated by an in vitro transcription reaction (BioArray HighYield RNA Transcript Labeling Kit; Enzo, Farmingdale, NY, USA). The fragmented cRNA was hybridised to the murine U74Av2 GeneChip (Affymetrix, High Wycombe, UK) representing 12,500 sequences (functionally characterised sequences and expressed sequence tag clusters). The washing procedure was carried out using the GeneChip Fluidics Station (Affymetrix) according to the manufacturer's protocol. The hybridised cRNA was stained with r-phycoerythrin-streptavidin (Molecular Probes, Karlsruhe, Germany) followed by an antibody amplification procedure with a biotinylated anti-streptavidin antibody (Vector Laboratories, Burlingame, CA, USA). The chips were scanned with a GeneArray Scanner (Hewlett Packard, Böblingen, Germany). Data were analysed with the Affymetrix Microarray Suite (MAS 5.0) and Microsoft Excel software. The expression level of a single mRNA was determined as the average fluorescence intensity among the intensities obtained by 16-paired (perfect-matched and single nucleotide-mismatched) probes consisting of 25-mer oligonucleotides. To identify differentially expressed genes, genes that were scored absent in the test sample (upregulated genes) or absent in the control sample (downregulated genes) were excluded. Genes that were differentially expressed between the two clean air exposure times (4 and 24 h) were only included if the comparison of the test sample to both control samples revealed a two-fold or more change. The given fold changes for genes with a difference call increase or decrease were used.
RT-PCR and real-time RT-PCR
For RT-PCR and real-time RT-PCR, 1 µg of total RNA was used for the first-strand cDNA reaction using hexamer primer (Promega Corporation, Mannheim, Germany) and Superscript II reverse transcriptase (Invitrogen Life Technologies) at 42°C for 50 min. PCR was performed on aliquots of this reaction in a total volume of 25 µL. For real-time PCR, the first-strand cDNA template, primer mix (as for RT-PCR) and SYBR Green PCR Master Mix (Applied Biosystems) were used in a total volume of 20 µL. Primers for ribosomal 18S mRNA were used as a control for each template in every experiment. The reactions were repeated with independently isolated RNA samples from a single animal per experimental condition. Expression of target genes was normalised to ribosomal 18S mRNA and displayed as fold-change relative to the sample from the control animals. The experiments were performed with the ABI Prism 7000 SDS (Applied Biosystems).
In situ hybridisation
In situ hybridisation was performed with a single-stranded digoxigenin-labelled RNA probe on paraffin-embedded lung sections as described elsewhere 16. For each experimental condition, two mice were analysed. Sense and antisense probes specific for osteopontin, galectin-3 and lipocalin-2 were generated by RT-PCR and in vitro transcription.
Histology and immunohistochemistry
Lungs from exposed and control mice were fixed in buffered formalin at an inflation pressure of 20 cmH2O and embedded in paraffin. Slides from exposed and control lung tissues were stained with polyclonal antibodies against galactin-3 (Cedarlane Laboratories Ltd, Hornby, Ontario, Canada), osteopontin (R&D systems, Wiesbaden, Germany) and lipocalin-2 (R&D systems). After staining with a biotinylated secondary antibody (Vector laboratories Inc.) and streptavidin-Vectastain Elite ABC-peroxidase reagents (Vector Laboratories Inc.), slides were developed with diaminobenzidine (Vector Laboratories Inc.). Negative controls tissues were stained without primary antibody.
Analysis of protein secretion
Per assay, 50 µL of cell-free BALF were applied. Mouse-specific enzyme-linked immunosorbent assays for osteopontin (Assay Designs, Ann Arbor MI, USA), keratinocyte-derived chemokine (KC), tumour necrosis factor (TNF)-α, interleukin (IL)-10, IL-12p40 and IL-1β (R&D Systems) were used according to the manufacturer's instructions.
Statistical analyses
Values are reported as mean±se. ANOVA was used to establish the statistical significance between the different experimental groups. Tukey's honestly significant difference procedure was applied to differentiate significant differences between the groups. Differences were considered significant at p<0.05.
RESULTS
Characteristics of particles
UFCPs showed a monomodal number distribution with a median particle size (equivalent mobility diameter) of 48.9±1.8 nm, mean geometric sd of 1.53, and mean number concentration of 7.7±0.8×106 cm-3. The average UFCP mass concentration in the exposure chamber was measured as 380 µg·m-3 (fig. 1⇓). According to the particle spectra, 92.3% of the generated particles were classified as ultrafine and 7.7% as fine. It was estimated that 85% of the deposited mass in rodent lungs was from UFPs and 15% from fine particles. This estimation was carried out using the multiple-path particle deposition model 17, assuming standard breathing conditions. The particle mass distribution needed for this calculation was derived from the number distribution reported above and a particle density varying with size, calculated according to Naumann 18.

Average number size distribution of 77 samples of generated ultrafine carbon particles (UFCP) in 24 h. UFCPs show a median particle size of 48.9±1.8 nm and mean number concentration of 7.7±0.8×106 cm-3. Of the generated particles, 92.3% are classified as ultra-fine (<0.1 µm).
BALF cell and protein parameters after inhalation of UFCP
After UFCP or clean air inhalation for 4 and 24 h, mice were sacrificed and BAL was performed to analyse cellular distribution and protein levels. The total number (0.45±0.05×106) of lavaged cells was unchanged after UFCP inhalation (fig. 2a⇓). Noticeably, the fraction of polymorphonuclear cells (PMN) appeared slightly, but not significantly increased after particle inhalation for 24 h (fig. 2b⇓). The number of macrophages in BALF was not increased after UFCP inhalation (fig. 2c⇓). LDH, a marker for cytotoxicity, was not altered, but total protein concentration in BALF was significantly increased after 24 h of particle inhalation (fig. 2d⇓). This, together with the slightly elevated PMN number, points to a very mild inflammatory response within the lungs after UFCP inhalation for 24 h.

Results of ultrafine carbon particle (UFCP) inhalation for 4 and 24 h in a) total cell number, b) polymorphonuclear cells (PMN), c) macrophages, and d) protein in bronchoalveolar lavage fluid. □: control group; ░: UFCP exposed subjects. Data are presented as mean±se from eight animals. *: p<0.05.
Changes in gene expression after inhalation of UFCPs for 4 h
According to analysis of the hybridisation signals (MAS5.0 software; Affymetrix), between 45.6% (4-h clean air) and 47.2% (24-h clean air, 24-h UFCP inhalation) of 12,422 transcripts represented on the chip were detected (detection: p<0.04; signal: >30).
Among the genes that were significantly expressed (detection p-value <0.04; signal >30) and showed significant changes (p<0.003) according to the Affymetrix MAS5.0 software, the current authors identified 157 (1.3%) genes that were upregulated after 4 h of inhalation of UFP and 125 (1%) downregulated genes. From these genes, seven were induced two-fold or more, whereas no gene was more than two-fold downregulated (table 1⇓). Two genes, granzyme A and a 5′–3′ exonuclease, were 1.8-fold downregulated.
Changes in gene expression after inhalation of ultrafine particles for 4 h and 24 h
From the two-fold and more upregulated genes, five were heat shock proteins (hsp; table 1⇑). The highest induction, four-fold, was detected for hspa1A (the mouse homologue of hsp70), a hsp described to interact with apoptosis-inducing factor. The other upregulated genes with chaperone activity were hsp105 (2.6-fold induction), suppression of tumorigenicity-13 (2.2-fold induction), stress-induced phosphoprotein-1 (two-fold induction) and osmotic stress protein 94 (two-fold induction). A 2.2-fold induction was detected for carcinoembryonic antigen-related cell adhesion molecule-2.
Furthermore, the mRNA for prolyl-4-hydoxylase alpha (I)-subunit, a key enzyme in the biosynthesis of collagens was upregulated two-fold. Another eight genes fulfilled the criteria for an increase of expression (between 1.5-fold and two-fold induction). Amongst them WEre hsp47 and hsp40 and two molecules associated with electron transport, cytochrome b-561 and PFTAIRE protein kinase-1.
Changes in gene expression after inhalation of UFP for 24 h
After UFP inhalation for 24 h, expression of 236 (1.9%) genes was increased and 307 (2.5%) genes were decreased compared with the two clean air controls (detection p-value <0.04; signal >30; p<0.003) according to the Affymetrix MAS5.0 software. From these affected genes, 17 were induced and six repressed two-fold or more (table 1⇑). Noticeably, all two-fold or more upregulated genes were related to inflammatory processes. In particular osteopontin, lipocalin-2 (24p3) and galectin-3 have been implicated to be important mediators of inflammation and to play a role as an integrated part of the body’s defence system 19–21.
The highest increase, a 5.4-fold change, was detected for serum amyloid A3, a major acute-phase response protein secreted by activated macrophages. A 3.8-fold induction was detected for the mRNA of the tumour growth factor-β member activin B. Although the expression level was comparatively low, a three-fold increase was detected for prostaglandin-endoperoxide synthase 1 (cyclooxygenase-1). The mRNA expression of leucine-rich α-2-glycoprotein was 2.8-fold increased. Tissue inhibitor of metalloproteinase (TIMP)-1, another factor involved in extracellular matrix remodelling, appeared 2.4-fold upregulated. Expression of two protease inhibitors was 2.4-fold and 2.2-fold increased: serine protease inhibitor-2, representing the homologue of human α-antichymotrypsin and the protein kinase-120 precursor, a member of the inter-α-trypsin inhibitor superfamily. Additionally, the later two genes that are involved in acute phase response and blood coagulation, the mRNA for coagulation factor III (tissue factor), was 2.2-fold upregulated. Another gene involved in coagulation, thrombospondin-1, an extracellular matrix protein that is secreted by different cell types, including endothelial cells, fibroblasts, smooth muscle cells and type II pneumocytes, was 2.4-fold induced by UFCP exposure. A two-fold induction was detected for keratin complex-1 gene 19, a specific cytoskeletal component of simple epithelia, including bronchial epithelial cells. Another 11 genes were less than two-fold but >1.5-fold induced (data not shown), amongst them the extracellular matrix proteins tenascin-C and TIMP-2 and the suppressor of cytokine signalling-3.
Molecules repressed by UFCP inhalation were cytochrome P1-450, receptor-type protein tyrosine phosphatase, translation initiation factor-2C, nuclear factor I/C, ryanodine receptor RyR1 and E-selectin ligand-1. To validate the results of the microarray experiments, the current authors performed real-time RT-PCR on lung RNA from single animals (fig. 3⇓). For all genes examined, the results obtained were confirmed by microarray hybridisation. The changes were in the same order of magnitude as detected by expression profiling.

Validation of differential gene expression induced by inhalation of ultrafine carbon particles (UFCPs) by real-time RT-PCR. BALB/cJ mice were exposed to UFCP for 4 h (□) and 24 h (░). RNA preparation and RT-PCR were carried out as described in the Materials and Methods section. Data are presented as the mean of eight samples per experimental group.
Inhalation of UFP leads to macrophage activation
Several genes, e.g. cytokines and cell adhesion molecules, which were found to be upregulated in the present expression study, are known to be involved in inflammatory cell activation. To analyse their expression and cellular distribution in the lung, exemplary galectin-3, osteopontin and 24p3 were chosen for in situ hybridisation experiments. Lungs of mice exposed for 24 h to either UFCP or clean air were compared.
In lungs from clean air control animals, galectin-3 expression was hard to detect by in situ hybridisation. Only a few single alveolar epithelial cells were weakly labelled (fig. 4⇓). After inhalation of UFCP for 24 h, a positive staining of alveolar macrophages and alveolar epithelial cells was clearly recognisable. No signal was detected after hybridisation with the galectin-3 sense probe.

Osteopontin (a, d, g and j), galectin-3 (b, e, h and k) and lipocalin-2 (c, f, i and l) were upregulated after inhalation of ultrafine carbon particles (UFCPs). Paraffin-embedded lung sections of control animals (a–c) and animals after 24-h UFCP inhalation (d–l) were hybridised to Dig-labelled antisense probes (a–i) or antibodies (j–l). In situ hybridisation with an osteopontin antisense probe revealed almost no staining in control sections; however, after inhalation, alveolar macrophages (AM) showed a positive signal (d and g). This result was confirmed by immunohistochemistry with a polyclonal osteopontin antibody (j). According to in situ hybridisation (e and h) and immunohistochemistry (k), galectin-3 is upregulated after UFCP particle inhalation in AM. Lipocalin-2 expression was not detected in control animals by in situ hybridisation (c). After particle inhalation, a signal was detected by in situ hybridisation (f and i) and immunohistochemistry (l) in lung septal cells (SC). AS: alveolar space. Scale bar = 50 μm (a–f), 25 μm (g–i) and 10 μm (j–l).
In situ hybridisation experiments with an osteopontin-specific antisense probe on mouse lung revealed positive staining of alveolar macrophages after 24-h inhalation of UFCP (fig. 4⇑). In lungs from clean air control mice, after hybridisation with the sense probe, no staining was detected (fig. 4⇑).
Lipocalin-2 (24p3) expression was detected in alveolar wall epithelial cells from lungs after 24-h UFCP inhalation (fig. 4⇑). No staining was seen in lung sections from clean air controls and after hybridisation with the sense probe.
These results were confirmed by immunohistochemistry. In alveolar macrophages of control animals, a slight staining was detected by anti-osteopontin and anti-galectin-3 antibodies. This signal increased after UFCP particle inhalation (24 h). Both antibodies revealed a staining in bronchiolar cells. However, this signal did not increase after UFCP inhalation. Lipocalin-2 protein expression was detected in lung septal cells of control animals. This signal was increased after particle inhalation (fig. 4⇑).
Secretion of osteopontin into BALF after inhalation of UFCPs
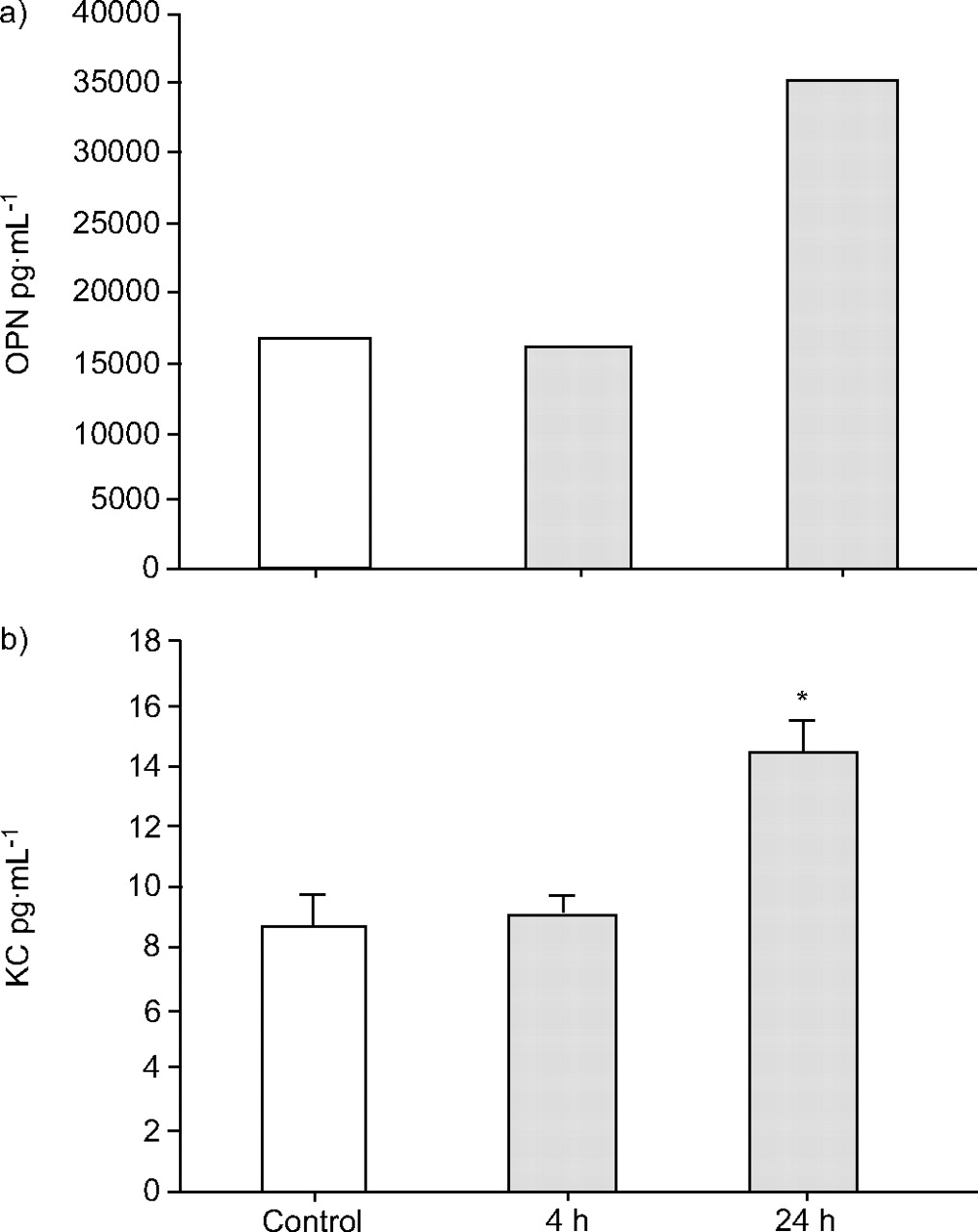
Osteopontin is a secreted protein, which is found in extracellular fluid and serum. To determine whether newly expressed osteopontin protein is enriched in BALF, an osteopontin-specific immunoassay with BALF samples from UFCP- and clean air-exposed mice was performed (fig. 5⇓). BALF samples from eight animals of each experimental group were pooled. In clean air control, 18 ng·mL-1 osteopontin protein was detected. After UFCP inhalation for 24 h, osteopontin protein concentration increased to 35 ng·mL-1. In concordance with the current authors’ chip and in situ mRNA expression data, osteopontin protein is about two-fold enriched in BALF from animals after 24-h UFCP inhalation. No increase could be observed after UFCP inhalation for 4 h.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
a) Osteopontin (OPN) and b) keratinocyte-derived chemokine (KC) concentration in bronchoalveolar lavage fluid (BALF) after particle inhalation. Osteopontin levels in BALF pools (eight animals per experimental group) and KC levels in unpooled BALF samples were measured by ELISA. Inhalation of ultrafine carbon particles (UFCPs) increases osteopontin concentration from 18 ng·mL-1 (control) to 35 ng·mL-1 (24-h UFCP). An increase in KC concentration from 9 pg·mL-1 to 15 pg·mL-1 was measured after 24-h particle inhalation. Data are presented as the mean±se of eight separate experiments. *: p<0.05.
For further characterisation of the inflammatory response to UFCP exposition, the secretion of cytokines and chemokines, known as inflammatory markers, into BALF was examined. KC, the mouse homologue of IL-8, can be detected in BALF by ELISA. KC protein levels in BALF from control animals and after 4-h particle inhalation did not differ. After particle inhalation for 24 h, KC concentration increased significantly from 9.0±0.5 to 14.4±0.8 pg·mL-1 (fig. 5⇑). TNF-α could not be detected in BALF. IL-1β showed a mild but not significant increase from 1.2±0.2 to 1.5±0.2 pg·mL-1 after 24-h UFCP inhalation. IL-10 protein levels decreased from 0.7±0.2 to 0.4±0.2 pg·mL-1, again not statistically significant, while IL-12p40 increased from 4.1±0.4 to 5.2±0.3 pg·mL-1.
DISCUSSION
UFP have been identified in epidemiological studies as an important factor inducing adverse health effects, such as cardiovascular complications and aggravation of respiratory diseases 1–3. In animal experiments, intratracheal instillation of UFP beyond a certain mass and surface area dose has been shown to cause acute pulmonary inflammation 22, 23. Exposures of healthy and asthmatic subjects to 25 µg·m-3 UFCP did not cause any detectable changes in airway inflammation 15; however, they caused alterations in the expression pattern of adhesion molecules on blood cells, indicating increased retention of leukocytes in the pulmonary vascular bed 24. The present study provides evidence that inhalation of elemental carbon particles at doses >40-fold above environmental relevant doses induces mild pro-inflammatory processes within 24 h of exposure. Beyond this, a comprehensive expression profiling approach gives an insight into early pathways leading to these processes.
Oxidative stress, caused by ambient particles deposited in the lung, is believed to be the main factor driving inflammatory and noxious effects 25. Even though the mechanism of the generation of oxidative stress is not understood, it appears to be related to the surface properties and the large particle surface area of UFP. Thus, cell–particle interactions in the lung might lead to the activation of pro-inflammatory gene transcription via the induction of nuclear import of redox-sensitive transcription factors, such as nuclear factor (NF)-κβ and activator protein-1. Several studies suggest that the particle-bound nonelemental carbon impurities account for the surface reactivity of UFP. Li et al. 26 and Xia et al. 27 observed a direct correlation between oxidative stress and the organic carbon content of UFP, in particular the polycyclic aromatic hydrocarbons and quinones. Dick et al. 28 showed that the property of UFP to cause oxidant damage is related to their ability to generate surface free radicals. In this process, transition metals may be involved via Fenton chemistry 29.
To mimic ambient conditions, the use of ambient UFP should be the most realistic exposure. However, apart from problems of nonreproducibility due to varying compositional changes in ambient particle samples and the bias arising from sampling artefacts, the aerosolisation of collected particles at the ultrafine range is, at present, very problematic. Like combustion-derived UFPs, which represent the major component of urban UFPs, the PALAS-generated soot particles (UFCP) that were used in the present study represent carbonaceous nanoparticles. Although urban particles contain trace amounts of organic compounds and metals, the current study focused on the effects of pure carbon particles.
The essential advantage of laboratory-made UFCPs is the possibility to produce defined spectra of particle sizes with similar surface and organic content properties. Thus, UFCPs are produced with organic mass contributions <5% 14, 15. PALAS soot particles are known to have similar physical properties like freshly generated diesel soot, e.g. an electron spin resonance-signal characteristic for organic carbon-centred radicals 30, 31. This feature might be important for hydroxyl generation and downstream effects on cellular oxidative stress 32. Recently, Beck-Speier et al. 31 demonstrated the high oxidative potential of UFCP in a cell-free system, as well as the production of the oxidative stress marker 8-isoprostane in exposed alveolar macrophages.
In the present study, a UFP concentration of 380 µg·m-3 was used. This is 10–100-fold higher than ambient levels measured in polluted urban areas 8. This mass concentration is in the range (170–1660 µg·m-3) applied in other toxicological studies identifying only mild inflammatory responses to UFP exposure 33, 34. The pro-inflammatory effects described in the current study could also be detected less pronounced at a lower particle burden of 180 µg·m-3, e.g. the release of osteopontin in BALF increased 1.5-fold after 24-h inhalation of 180 µg·m-3 UFCPs. Although this mass concentration is still high in comparison to ambient UFP measured at urban sites, e.g. in Munich (Luise-Kiesselbach-Platz, autumn 1998, 8 µg·m-3; E. Karg, GSF-National Research Center for Environment and Health, Neuherberg, Germany, personal communication), it should be notified that the particle effects described were already detectable after acute exposures and have been found in young and healthy animals. Aged and compromised animals are expected to have a stronger response to particle exposure. According to this, Elder et al. 35 exposed different groups of healthy and compromised mice to 110 µg·m-3 UFCP for 6 h and found significant lung inflammatory response only in aged, emphysematous animals, but not in healthy mice.
In the present study, overall histology of the lung did not give any indication for inflammation or pathological changes. Examination of BALF showed a small increase in PMN cell number after UFCP inhalation (4.0±1.2×103) compared with clean air controls (2.6±0.5×103), suggesting a very mild inflammatory cellular response (fig. 2⇑). This was reinforced by a moderate but significant increase in BALF protein concentration after 24 h of UFCP inhalation. These results fit into a recently published study by Frampton et al. 15 where healthy and mildly asthmatic volunteers exposed for 2 h to UFCP (10 µg·m-3 and 25 µg·m-3) did not reveal any significant effects on pulmonary functions or markers for airway inflammation.
However, DNA microarray profile analysis already revealed a remarkable upregulation of mRNA expression after 4 h of UFP inhalation. Five out of seven significantly induced genes encode hsp and proteins with chaperone activity. This induction is transient and no longer detected after UFCP inhalation for 24 h. Based on their ability to chaperone antigenic peptides, hsp can elicit specific cellular adaptive immune responses 36. These immunmodulatory properties of hsp are likely to be responsible for the initiation of the immune response after particle inhalation. The current results support this hypothesis, as they revealed the subsequent induction (after 24-h UFP inhalation) of several genes known to be regulated via the NF-κB signalling pathway. Amongst them are galectin-3 and lipocalin-2 (24p3), whereas osteopontin has recently been shown to activate NF-κB via induction of induced-κB phosphorylation and degradation through inhibitor of NF-κβ kinase. Additionally, the current authors noted transiently increased transcript levels of the alpha (I)-subunit of prolyl-4-hydoxylase after 4 h of particle inhalation. Prolyl-4-hydoxylase is a key enzyme in the biosynthesis of collagens. Collagen synthesis is increased during connective tissue remodelling that occurs in allergic asthma. In accordance to the induction of hsp70 by UFCP exposure that is reported in the present findings, it was recently shown that exposure of human alveolar epithelial cells to ultrafine carbon black particles induced hsp70 as a result of oxidative stress 37.
The highest induction after UFCP exposure for 24 h was detected for serum amyloid A (SAA)-3, the predominant SAA isoform expressed extrahepatically. SAA3 is known to be secreted by macrophages after lipopolysaccharide (LPS) treatment. The upregulation of SAA3 after particle inhalation indicates an inflammatory response, although the increase is not as high as after LPS induction.
According to the current results, osteopontin seems to play a central role in the induction of pro-inflammatory processes by UFP inhalation. Osteopontin is a secreted and glycosylated phosphoprotein that contains the arginine-glycine-aspartic acid (RGD) integrin-binding domain. Osteopontin protein has chemokine/cytokine-like properties and is among the most abundantly expressed proteins in a wide range of lung diseases, such as fibrosis, sarcoidosis and lung carcinoma 19, 38. In the present study, in situ hybridisation experiments revealed upregulation of osteopontin expression in alveolar macrophages and a two-fold increase of secreted osteopontin protein was detected by ELISA in BALF after UFP inhalation. In accordance with the current in situ results, immunohistochemistry revealed osteopontin protein expression in alveolar macrophages.
Recently, it was shown that osteopontin is implicated in experimental particle-induced lung disease using a titanium dioxide exposure model in a rat. Under exposure conditions, which resulted in fibroproliferative lung disease (long-term exposures at particle concentrations >10 mg·m-3), rats had significant increases in total lung osteopontin mRNA expression and increased levels of osteopontin protein in BALF prior to the development of lesions 40. Osteopontin is secreted by activated macrophages, leukocytes and activated T-lymphocytes, and is present in extracellular fluid at sites of inflammation, and in the extracellular matrix of mineralised tissues. In the immune system, osteopontin plays a role in chemotaxis, leading to the migration of macrophages and dendritic cells to sites of inflammation. Osteopontin protein interacts with a variety of cell surface receptors, including the αvβ3, αvβ1, α41β, α8β1 and α9β1 integrins, as well as CD44. Binding of osteopontin protein to these cell surface receptors stimulates cell adhesion, migration, and specific signalling functions. The major integrin-binding site of osteopontin is the RGD integrin-binding motif, which is required for the adherence of many cell types. Ashkar et al. 39 have demonstrated a differential regulation of macrophage IL-12 and IL-10 expression by osteopontin, which affects type-1 immunity. Interaction between osteopontin protein and macrophages is mediated through αvβ3 integrin and CD44. The phosphorylation-dependent interaction between osteopontin protein and its integrin receptor stimulates IL-12 expression, whereas the phosphorylation-independent interaction with CD44 inhibits IL-10 expression by macrophages. In the present study, IL-10 protein concentration was slightly diminished in BALF after UFCP inhalation, as was demonstrated by ELISA. Moreover, IL-12 concentration was raised slightly but not significantly after particle inhalation.
To support the current hypothesis that UFP inhalation triggers inflammatory processes, the expression of other known markers for inflammation in relation to particle inhalation was examined. A significant increase of KC protein concentration in BALF was detected, although the mRNA expression was not upregulated. KC, a CXC chemokine, is released by activated macrophages and injured epithelial cells and acts as a chemoattractant for neutrophils. It has also been implicated in the accelerated release of neutrophils in response to inflammation. In the lung, the release of matrix-bound KC by shedding is important for the migration of neutrophils from the interstitium to the alveolar space 41.
Lipocalin-2 (24p3, neutrophil gelatinase-associated lipocalin (NGAL)) is upregulated after 24-h UFP inhalation in epithelial cells of the alveolar wall. Lipocalins are small secreted proteins that play a role in diverse biological processes through binding of small hydrophobic molecules, encompassing retinoids, fatty acids, prostaglandins and odorants 20. Lipocalin-2 expression is induced by different stimuli in diverse tissues and cell lines. It was identified as a LPS-induced protein secreted by mouse macrophages 42. Recently, it was demonstrated that lipocalin-2 was expressed in bronchial goblet cells as well as in alveolar type II pneumocytes, and that the expression is increased in bronchial and alveolar cells of inflamed human lungs 42. The increase of lipocalin-2 mRNA expression is induced by IL-1β via a NF-κB-dependent pathway in the human type II pneumocyte-derived cell line A549. No induction of lipocalin-2 transcript was seen in A549 cells after stimulation with LPS, TNF-α and IL-6. In the mouse lung, liocalin-2 was detected by in situ hybridisation in septal cells after particle inhalation (fig. 4⇑). No hybridisation signal was detected in bronchial cells and in control animals. It remains to be investigated in further in vitro experiments if the lipocalin-2 expression in alveolar epithelial cells is upregulated directly by UFPs that are supposed to enter these cells because of their small size 11, 43, or if the increased expression and secretion of a cytokine, such as IL-1β, by alveolar macrophages is necessary for lipocalin-2 upregulation. Even if the function of lipocalin-2 is not known, increased serum levels of the human homologue NGAL have been related to the clinical manifestation of cardiovascular disease 44. Recently, a putative function of this lipocalin as a modulator of the inflammatory response has been suggested. Under normal circumstances, granulocytes have a short life span and die by apoptosis. In many chronic inflammatory responses, such as bronchial asthma or lung fibrosis, delayed apoptosis of granulocytes leads to their accumulation at sites of inflammation, where they cause tissue damage via the release of toxic mediators 45.
Interestingly, osteopontin and lipocalin-2 seem to play a role in the pathogenesis of atherosclerosis 44, 46. People suffering from this cardiovascular disease are one of the groups considered particularly susceptible to the effects of airborne particles.
Galectin-3 is a β-galactoside-binding lectin, implicated in inflammatory responses as well as in cell adhesion. The role of galectin-3 as an adhesion molecule for neutrophil extravasation during streptococcal pneumonia has been recently demonstrated 47. Recently, the contribution of galectin-3 to phagocytosis by macrophages has been shown 48. Galectins, in particular galectin-1 and galectin-3, play a role as regulators of inflammatory processes. It was demonstrated that alveolar infection with Streptococcus pneumoniae, but not with Escherichia coli, induces the production and secretion of galectin-3 by alveolar macrophages 48. Accumulation of galectin-3 in the alveolar space of streptococcus-infected lungs correlates with the onset of neutrophil extravasation. In vitro assays revealed the ability of galectin-3 to mediate the adhesion of neutrophils to endothelial cells. These data suggest that galectin-3 is implicated in β2 integrin-independent neutrophil extravasation induced by S. pneumoniae; however, not in β2 integrin-dependent pulmonary infection induced by E. coli. Induction of galectin-3 expression in alveolar macrophages by UFP inhalation might maintain neutrophil extravasation in a similar way.
In conclusion, the current data provide evidence that inhalation of ultrafine carbon particles triggers a biphasic pro-inflammatory process in the lungs of healthy mice. After a short exposure to ultrafine particles, heat shock proteins are transiently upregulated and might be responsible for the subsequent activation of macrophages. The present study demonstrates that inhalation of ultrafine carbon particles induces the upregulation of osteopontin and galectin-3 expression in alveolar macrophages. Osteopontin, galectin-3 and lipocalin-2 are secreted proteins and their regulation might serve as a useful marker for inflammatory processes induced by particle inhalation in further studies.
Acknowledgments
The authors would like to thank G. Ferron and O. Schmid for help with the application of the rodent deposition model and critical reading of the manuscript.
- Received June 16, 2005.
- Accepted April 20, 2006.
- © ERS Journals Ltd
References










