





Abstract
The central role of lung ischaemia–reperfusion injury in pulmonary dysfunction after cardiac surgery, particularly thoracic organ transplantation, has been well recognised.
Lung tissue necrosis after prolonged ischaemia is known to worsen lung function, which was believed to be due largely to adjacent tissue inflammation. Recent studies suggest that lung apoptosis following ischaemia–reperfusion could be equally important in the development of post-operative lung dysfunction.
The current literature on the mechanism and pathways involved in pulmonary dysfunction and, in particular, its relationship with apoptosis after lung ischaemia–reperfusion is briefly reviewed here.
A better understanding of lung apoptosis, as well as the upstream pathways, may help in the development of therapeutic strategies that could benefit patients undergoing cardiac and lung transplantation.
Ischaemia–reperfusion-induced pulmonary dysfunction is a significant clinical problem in cardiac surgery and, particularly, lung transplantation 1. Post-operative pulmonary dysfunction following the use of cardiopulmonary bypass is a frequently observed phenomenon that is associated with lung ischaemia–reperfusion injury 2, 3. Potentially life-threatening graft dysfunction early after lung transplantation can occur in up to 20% of patients 1. However, understanding of the complex pathophysiology of ischaemia–reperfusion-induced lung injury remains incomplete. Unlike any other organ in the human body, the lung possesses two blood supply networks with extensive anastomotic connections and a total of three potential sources of lung tissue oxygenation, thus making lung ischaemia–reperfusion injury more intriguing to study 2. Since the mid-1980s, the role of neutrophils, free radicals and other inflammatory mediators in ischaemia–reperfusion injury has been extensively investigated 4. Nevertheless, these mediators appear to contribute only in part to lung ischaemia–reperfusion injury. The final pathways, after lung ischaemia and reperfusion, leading to cellular damage, necrosis or apoptosis of the pulmonary epithelium remain to be fully elucidated. Recently, the mechanisms involved in pulmonary apoptosis following lung ischaemia–reperfusion have begun to be understood 5. The current review highlights the latest research findings in apoptosis after acute lung ischaemia–reperfusion injury.
IS APOPTOSIS INVOLVED IN LUNG ISCHAEMIA–REPERFUSION INJURY?
Pulmonary ischaemia–reperfusion can cause cellular breakdown and death of lung epithelial tissue, which may contribute to the magnitude and duration of pulmonary dysfunction seen after cardiopulmonary bypass and lung transplantation 2, 4. Since the late 1990s, scientists have recognised that the different processes following ischaemia–reperfusion, although closely related, cause lung injury by activation of different inflammatory pathways 2, 4, 6. Consequently, these changes may lead to pulmonary cellular necrosis or apoptosis. Tissue necrosis after lung ischaemia–reperfusion injury has been recognised to be associated with significantly worsened lung function, related to the high degree of inflammation. However, the complex relationship between ischaemia–reperfusion and lung apoptosis is only beginning to emerge.
Apoptosis, also known as programmed cell death, was reintroduced to modern clinical practice by Kerr et al. 7 in 1972, although the original medical use of the word could be traced back to the era of Hippocrates (c. 460–370 BC) and Galen (AD 129–201) 8. Apoptosis means “falling off or dropping off” in Greek, and the process is fundamental to normal organ development. Morphologically, apoptosis is characterised by nuclear shrinkage, chromatin condensation and DNA fragmentation to oligonucleosome-sized fragments, whereas plasma membrane and intracellular organelles remain intact 9. Recent evidence indicates that apoptosis may also play an important role in pulmonary disease processes such as lung cancer, interstitial pulmonary fibrosis and adult respiratory distress syndrome (ARDS) 10, 11. There are many factors that comprise the normal and abnormal regulation of cell death leading to apoptosis in pulmonary epithelial cells, which are likely to be different in each unique situation 5.
In experimental studies of rodent single-lung transplantation, a short duration of ischaemia (20 min) and reperfusion was associated with neither increased cellular necrosis nor apoptosis of the transplanted lung compared with the pre-retrieval lungs 12. After prolonged periods (up to 18 h) of cold ischaemic lung preservation without reperfusion, a high percentage of cell necrosis (but not apoptosis) can be observed 12, 13. Reperfusion of lung epithelial cells is known to increase the degree of damage when the transplanted lung is subjected to a long period of cold ischaemia, since the amount of cellular necrosis and apoptosis is significantly influenced by the ischaemic duration. For example, following a moderate duration (6 or 12 h) of cold ischaemic preservation, the mode of pulmonary epithelial cell death in the transplanted lung upon reperfusion was dominated by apoptosis (<2% necrosis, 30% apoptotic). In contrast, after longer periods of cold ischaemia lasting 18 or 24 h, the mode of such cellular death was necrosis dominant (21–29% necrosis, <1% apoptosis) 12. Similarly, in clinical lung transplantation, almost no apoptosis was detected after cold or even warm ischaemia alone for a duration of up to 5 h 14. Apoptosis at the alveolar epithelium only became evident following reperfusion, and, furthermore, the number of apoptotic cells increased with longer periods of reperfusion 14. Ischaemic lung pneumocyte apoptosis was found to peak at 2 h after reperfusion, subsequently decreasing with prolonged reperfusion 13. In addition, apoptosis was independent of the total ischaemic time, preservation solution or ventilation time prior to lung harvesting 14. Therefore, lung ischaemia alone under cold and warm conditions, or reperfusion of the lung after only a short period of cold ischaemia, appears to have minimal effects on lung apoptosis. However, lung reperfusion after long periods of ischaemia leads to significant apoptosis early after reperfusion, as well as necrosis when the ischaemic time is further prolonged. The accepted theory for reperfusion lung injury is primarily based on the generation of reactive oxygen species (ROS) upon reperfusion, causing cellular damage and apoptosis. Another interesting concept is that apoptosis is an energy-dependent programmed process of cell death. Hence, the absence of apoptosis after prolonged ischaemia may simply be a reflection of depleted energy reserves. Moreover, prolonged lung ischaemia may shift the balance towards pulmonary epithelial cell necrosis rather than apoptosis through increased levels of anti-inflammatory cytokines, such as interleukin (IL)-10 15.
A direct correlation between the degree of lung apoptosis after lung ischaemia–reperfusion and pulmonary dysfunction remains controversial. Interestingly, the deterioration in lung function following ischaemia–reperfusion, including decreased arterial oxygen tension (Pa,O2), was only associated with the percentage of cell necrosis and not the amount of apoptosis 12, 15. The poorer lung function associated with more cellular necrosis may partly be related to the high degree of surrounding inflammation seen with necrotic cells. This phenomenon is absent with apoptosis because apoptotic cells are readily phagocytosed by macrophages, thereby limiting the release of inflammatory intracellular enzymes from apoptotic cells as cell membrane fragments 16. The percentage of apoptotic cells after reperfusion in human lung transplantation was found not to be associated with certain clinical parameters, such as Pa,O2, 30-day mortality or post-operative mechanical ventilation time 14. Similar findings were also observed in a rodent lung transplantation model in that there was no correlation between lung function (Pa,O2, wet:dry weight ratio and peak airway pressure) and the degree of apoptosis 17. Indeed, improved lung function could be related to reduced pro-inflammatory 17 but enhanced anti-inflammatory 15 cytokine responses. However, in an experimental study, there was significant correlation between the amount of apoptosis after lung ischaemia–reperfusion and pulmonary shunt fraction, as well as between apoptosis and cellular oxidative injury 18. Thus, human studies appear to confirm the data from animal models that apoptosis is primarily a post-reperfusion event, and the degree of cellular necrosis after ischaemia–reperfusion appears to show a stronger correlation with disturbance in lung function than with apoptosis 4, 12. Although many previous experimental studies have suggested the relationship between apoptosis and lung dysfunction, they were mainly performed in the context of ARDS and other forms of acute lung injury, and few were specifically related to ischaemia–reperfusion-induced lung injury 10.
MECHANISM OF APOPTOSIS IN LUNG ISCHAEMIA–REPERFUSION INJURY
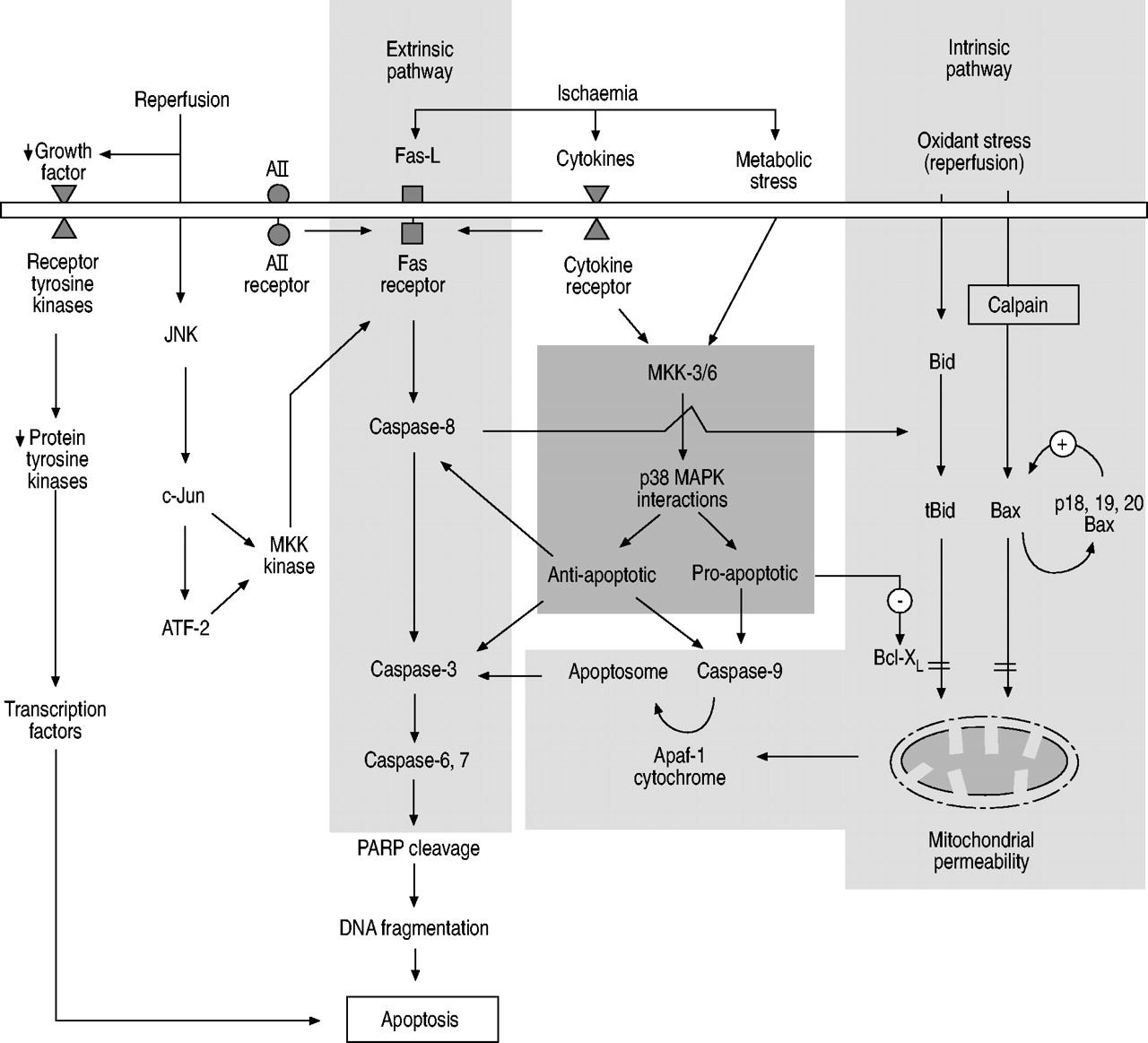
Apoptosis can be triggered by mechanical injury and exposure to certain environmental conditions leading to activation of the intrinsic (mitochondrial) and extrinsic (death receptor) pathways (fig. 1⇓). Activation of the extrinsic pathway by specific receptor–ligand interactions, such as Fas/Fas-ligand (Fas-L), angiotensin (A)II and tumour necrosis factor (TNF)/receptor, leads to subsequent activation of intracellular apoptotic cascades involving caspases (cysteine aspartyl proteases). The intrinsic pathway, known as the mitochondrial pathway, involves BH3-interacting domain death agonist (Bid) cleavage, mitochondrial membrane permeability, mitochondrial cytochrome c and apoptotic protease activating factor (Apaf) release, with apoptosome formation. In addition, levels of growth factors, including vascular endothelial growth factor, platelet-derived growth factor, epidermal growth factor and macrophage colony-stimulating factor, in the surrounding cellular environment can influence pulmonary epithelial cell apoptosis through intracellular protein tyrosine kinases and protein tyrosine phosphatases. These pathways may be downregulated by certain mitogen-activated protein kinases (MAPKs) and molecules from the B-cell leukaemia/lymphoma-2 gene product (Bcl-2) family. The phosphatidylinositol-3′-kinase and protein kinase B pathway has also been shown to be important in lung apoptosis. However, no studies have specifically explored the role of this pathway in lung ischaemia–reperfusion-induced apoptosis 19. The balance between these intracellular pro- and anti-apoptotic factors may ultimately determine whether a cell survives or descends down the pathway to undergo apoptosis. At present, the relative importance of each apoptotic factor and pathway in ischaemia–reperfusion lung injury remains to be elucidated. Furthermore, interactions of other external factors in the clinical setting with epithelial cell apoptosis (i.e. ventilator-induced lung epithelial cell apoptosis or necrosis), neutrophil activation and apoptosis, and mediators from the systemic inflammatory response, may also significantly influence the final outcome 10. The present review focuses on ischaemia–reperfusion-induced apoptosis in pulmonary parenchymal cells, rather than in the inflammatory cells (i.e. neutrophils and macrophages) of the lung. The regulation of apoptosis in these two cell populations could have different outcomes as regards the degree of pulmonary injury after ischaemia–reperfusion.

{kind=link}
The major intrinsic (mitochondrial) and extrinsic (death receptor) pathways regulating apoptosis following lung ischaemia–reperfusion injury. The important pathway affecting balance between anti- and pro-apoptotic mediators is also shown. AII: angiotensin II; Fas-L: Fas-ligand; JNK: c-Jun N-terminal kinase; MAPK: mitogen-activated protein kinase; MKK: MAPK kinase; ATF-2: activation of transcription factor 2; PARP: poly-ADP-ribose polymerase; Apaf: apoptotic protease activating factor; Bid: BH3-interacting domain death agonist; tBid: truncated Bid; Bax: Bcl-2-associated X-protein; Bcl-XL: Bcl-2-like 1. ↓: decrease; +: activation; -: inhibition; = : inhibition (block).
The extrinsic pathway
The Fas and caspase cascade, also known as the extrinsic pathway, is perhaps the most powerful pathway in the initiation of apoptosis. Fas (CD95) is a cell membrane receptor that can be found in a number of cell types, including alveolar epithelial cells. Its corresponding ligand Fas-L (CD95L) is a member cytokine of the TNF family and its interaction with the Fas receptor can trigger commencement of the pathway towards apoptosis, in particular by activating caspase-8. It has been shown that, following lung tissue ischaemia, the initiator caspases (caspase-8 followed by caspase-9) are activated in this manner 20. Subsequently, activation by proteolytic cleavage of executioner caspases (caspase-3, -6 and -7) allows attack of their cellular substrates within the apoptotic cascade 20, leading to activation of more downstream caspases and poly-ADP-ribose polymerase and subsequent DNA fragmentation. Furthermore, in the ischaemic lung, another important apoptotic pathway involves caspase-8 activation of the intrinsic pathway through Bid cleavage and mitochondrial cytochrome c release 20, 21, although the alternative direct extrinsic pathway through TNF and Fas death signalling is also possible under ischaemic conditions 21. Inhibition of any of the above caspases in lung ischaemia–reperfusion can attenuate pulmonary apoptosis 20.
It is known that, following acute lung injury or ARDS, Fas and Fas-L levels are significantly increased in the bronchoalveolar fluid and epithelial cells lining the alveolae 10, 22. This increase can be partly accounted for by local release in the lung of soluble Fas-L 22, 23 from activated pulmonary neutrophils and membrane-bound Fas-L release during neutrophil cell membrane lysis 23. The increase in pro-inflammatory cytokine levels associated with lung injury can prolong neutrophil lifespan by inhibiting neutrophil apoptosis and delay resolution of the inflammatory response, and may prolong Fas-L release 23. Interestingly, higher Fas levels were associated with unstable haemodynamics and multi-organ failure, as well as poor clinical outcome, in patients with lung injury 22. Ischaemia–reperfusion of the lung in the experimental setting has also been shown to cause increases in Fas and Fas-L expression, with associated apoptosis in lung tissue and pulmonary arterial endothelial cells 20. The depletion or antagonism of Fas by Fas-L antibody administration can significantly reduce apoptosis after lung ischaemia–reperfusion 20.
Role of mitogen-activated protein kinase
MAPK signalling pathways are known to play an important role in controlling cellular proliferation, differentiation and apoptosis. The direct and indirect activation of the various MAPK modules (including p38 and c-Jun N-terminal kinase (JNK), etc.) by extracellular stimuli have recently been neatly summarised 24, 25. With its three isoforms α, β, and γ, p38 MAPK is a member of the MAPK superfamily involved in cell membrane-to-nucleus signal transduction for cell cycle regulation and survival. Activation of p38 MAPK by MAPK kinase-3, -4, and -6 can occur in response to metabolic stress, cytokines and ischaemia. The level of p38 MAPK activity has been associated with both pro-apoptotic (by production of pro-inflammatory cytokines and Bid cleavage) and anti-apoptotic activities after lung ischaemia–reperfusion injury 20.
In a rodent lung ischaemia–reperfusion model, inhibition of p38 MAPK activation resulted in lower levels of the pro-inflammatory cytokine IL-1β, which was associated with less alveolar injury and pulmonary oedema, as well as better Pa,O2, after reperfusion 26. Furthermore, inhibition of p38 MAPK by its inhibitor, added into Euro-Collins solution during cold storage, in a canine lung transplantation model, significantly improved Pa,O2, alveolar–arterial oxygen tension difference, pulmonary vascular resistance and cardiac output, as well as reducing pulmonary oedema and neutrophil infiltration, after transplantation 27. The improved cardiopulmonary function was found to correspond to inhibition of p38 MAPK activation by its phosphorylation at 30 min after lung reperfusion 27, 28. Interestingly, in rodent iso- and allograft lung transplantation models, p38 MAPK activation was significantly reduced 2 h after reperfusion, suggesting p38 MAPK activation may also have some anti-apoptotic properties 28. Such inconsistencies in pro- and anti-apoptotic characteristics are probably caused by the activation of different or combinations of different MAPK kinase and p38 MAPK isoforms (α, β and γ) under the influence of various stimuli 20. For example, MAPK kinase-3 preferentially leads to activation of p38-α and -γ, whereas MAPK kinase-6 activates p38-α, -β and -γ. The activated isoform p38-α MAPK was shown to be responsible for anti-apoptotic effects in the animal model of lung ischaemia–reperfusion injury through inhibition of caspase-3, -8 and -9 20, possibly through induction of the cytoprotective haemoxygenase-1 gene 29. The effects of different combinations of p38 MAPK and MAPK kinase isoform activities on apoptosis after lung ischaemia–reperfusion injury remain to be elucidated.
JNK, another member of the MAPK family, is known to be involved in stress-induced apoptosis. In particular, oxidant stress, such as that found after reperfusion, may induce neutral sphingomyelinase activity, resulting in accumulation of the signalling molecule ceramide 30. Subsequently, ceramide can activate JNK, leading to phosphorylation of c-Jun and activation of transcription factor (ATF)-2 11. ATF-2 and c-Jun can bind, and turn on, a MAPK kinase kinase promoter, resulting in increased transcription of Fas-L. This may also be an important signalling mechanism for oxidant-induced lung epithelial apoptosis.
Role of tyrosine phosphorylation
Cellular tyrosine phosphorylation by tyrosine kinases is known to prevent apoptosis and promote cell survival in many cell types. The balance between the pro-apoptotic tyrosine phosphatase and anti-apoptotic tyrosine kinases is important in determining cell survival. After cold ischaemic preservation of human lung for 2–5 h, protein tyrosine phosphorylation activity increases, thereby favouring anti-apoptotic behaviour after lung implantation and before reperfusion 31. However, the tyrosine phosphorylation activity of the tyrosine kinases decreased significantly during the 2 h of lung reperfusion, before returning to basal levels, which may correlate with the development of apoptotic cell death early after reperfusion during experimental and human lung transplantation 28, 31. Interestingly, the activity of the pro-apoptotic protein tyrosine phosphatase was also reduced 2 h after reperfusion of the ischaemic lung in an experimental transplantation study 28. Hence, the balance between the activities of the pro-apoptotic tyrosine phosphatase and anti-apoptotic tyrosine kinase may be more important than their individual levels.
Role of angiotensin II
Classically, the renin–angiotensin system (RAS) has been viewed as an endocrine system in blood pressure regulation. However, recent evidence suggests that the RAS can be involved in regulation of apoptosis through paracrine and autocrine mechanisms 32. The local intrinsic RAS is expressed in the distal lung parenchyma and, through AII, plays a central role in the signalling of apoptosis in alveolar epithelial cells by interacting with epithelial AII receptors, necessary for Fas-mediated apoptosis 10, 11, 33. Increased levels of angiotensin-converting enzyme (ACE), which converts AI to AII, and AII have been found in bronchoalveolar fluid from patients after various forms of acute lung injury 10, 11. Inhibition of ACE in a rodent lung ischaemia–reperfusion model was shown to cause significantly better post-reperfusion Pa,O2 and peak airway pressure, and less pulmonary oedema than controls 34. The preservation of lung function by inhibition of ACE appears not to be neutrophil sequestration- or lipid peroxidation-related, suggesting that AII may adversely affect lung function after ischaemia–reperfusion via other pathways, such as Fas/Fas-L apoptosis 34.
Bcl-2 family (Bid/Bax/Bcl-XL)
The pro-apoptotic proteins Bid and Bcl-2-associated X-protein (Bax) within alveolar epithelial cells can be induced by oxidative stress 35, causing apoptosis through the intrinsic pathway. Proteolytic cleavage of Bid to give truncated Bid and Bax increases mitochondrial permeability through their binding to the permeability transition pore complex, leading to dissipation of the mitochondrial inner transmembrane potential and cytochrome c release through the outer mitochondrial membrane 35, 36. Furthermore, Bax may be cleaved by calpain into p18 and p19 Bax, which can provide positive feedback for the interaction between Bax and mitochondria 37. The released cytochrome c and Apaf-1 from the mitochondria, together with caspase-9, form an apoptosome to activate caspase-3 and the subsequent caspase cascade towards cell apoptosis. Interestingly, ischaemia and reperfusion of lung tissue had no effects on pro-apoptotic Bax expression 20.
Bcl-2-like 1 (Bcl-XL) belongs to the Bcl-2 family, which can prevent apoptosis induced by a variety of stimuli via caspase inhibition. In addition, Bcl-XL has been shown to prevent anoxia-induced apoptosis of lung epithelial cells 38. It acts downstream of Bid and its expression can inhibit all the apoptotic changes induced by activated Bid, hence ameliorating mitochondrial damage 21, reducing mitochondrial cytochrome c release and limiting caspase-9 activation 38. Furthermore, the effects of Bax on the mitochondrial membrane can be prevented by Bcl-XL 35. Ischaemia–reperfusion injury has been shown to reduce the levels of the anti-apoptotic proteins Bcl-2 and Bcl-XL, probably through the MAPK kinase-3/p38 MAPK pathway 20. The inhibitor of apoptosis proteins and second mitochondria-derived activator of caspases pathway has also been shown to be important in lung cancer apoptosis 39, 40. However, no studies to date have specifically explored the role of these pathways in lung ischaemia–reperfusion-induced apoptosis.
OTHER STIMULI IN ISCHAEMIA–REPERFUSION-INDUCED LUNG APOPTOSIS
Reactive oxygen species
The importance of ROS has long been recognised in ARDS and lung ischaemia–reperfusion injury 41. ROS may be generated from activated neutrophils through the reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase pathway 4, or from lung parenchymal cells after pulmonary ischaemia–reperfusion (hypoxia–reoxygenation). Ischaemia can prime the lung tissue, whereby reintroduction of molecular oxygen upon reperfusion results in the generation of toxic oxygen metabolites via the mitochondrial cytochrome P450 system 42. Associated with ATP degradation, hypoxia has been found to activate cellular p38 MAPK, leading to xanthine oxidase activation by phosphorylation, thereby, upon reperfusion (reintroduction of oxygen) of lung tissue, bringing about generation of ROS 43, 44. Furthermore, nitric oxide synthase (NOS) and the recently identified nonphagocytic cell membrane/cytosolic NADPH oxidase may also be involved in generating ROS as signalling molecules in apoptosis 44, 45. ROS may induce pulmonary cellular injury by: 1) directly increasing endothelial intracellular calcium levels and activating calcium-dependent proteases 46; and 2) acting as biochemical intermediates in apoptotic signalling by activating MAPK, leading to cytochrome c release and the subsequent poly-ADP-ribose polymerase cleavage 44, 45.
Nitric oxide
ROS, such as NO, are known to mediate cellular hypoxia–reoxygenation injury either directly or indirectly through generation of other oxidising mediators. At high concentrations, NO can inactivate numerous mitochondrial respiratory enzymes, including mitochondrial aconitase, nicotinamide adenine dinucleotide-ubiquinone oxidoreductase (complex 1) and succinate-ubiquinone reductase (complex 2), as well as cytochrome oxidase 47. In addition, hypoxic conditions can enhance the production of inducible NOS, which may increase oxidant radical byproducts, such as the peroxynitrite anion, and lead to ROS-type cellular injury 44. However, NO may also exert cytoprotective antioxidant effects and attenuate reoxygenation injury, particularly when derived from constitutive NOS. For instance, reduced constitutive NOS expression following anoxia–reoxygenation has been suggested to promote cellular apoptosis by increasing Fas and decreasing Bcl-2 expression 48.
Cytokines
The levels of some pro-inflammatory cytokines, such as TNF and IL-8, which are known to stimulate pulmonary neutrophil migration, activation and degranulation, have been shown to increase significantly in lung tissue and the systemic circulation after ischaemia and reperfusion of the lung 4, 49. In clinical lung transplantation, elevated IL-8 levels in lung tissue after reperfusion correlate significantly with low Pa,O2, high airway pressure and the Acute Physiology and Chronic Health Evaluation score, which may lead to longer intensive care unit stay and higher post-operative mortality 49. The increased local release of TNF after ischaemia–reperfusion lung injury may induce apoptosis through JNK activation, which, in turn, promotes caspase-8 and Bid activity, and ultimately results in pulmonary dysfunction 50.
The anti-inflammatory cytokine IL-10 has been found to protect the lungs from ischaemia–reperfusion injury by inhibition of the inflammatory and T-cell-mediated immune response. Endogenous IL-10 can reduce pulmonary vascular fibrin deposition and microvascular thrombosis after lung ischaemia–reperfusion through balancing the activities of plasma plasminogen activator inhibitor and tissue plasminogen activator, and, in so doing, also attenuates the release of IL-1 51. In a rodent lung ischaemia–reperfusion model, IL-10 can limit lung reperfusion injury by attenuating pulmonary vascular permeability, possibly through the suppression of alveolar macrophage TNF-α production 52.
The interaction of pro- and anti-inflammatory cytokines with lung function after ischaemia–reperfusion lung injury has been well studied. However, the influence of the cytokine network on apoptosis after ischaemia–reperfusion lung injury is much less understood. In an IL-10 gene transfection model, IL-10 ameliorates lung injury after lung transplantation by modifying the mode of cell death in favour of apoptosis, rather than necrosis 15. Nevertheless, in another lung transplantation model, higher levels of IL-10 and lower levels of TNF-α did not correlate with the number of apoptotic cells or level of Bcl-2 expression in lung tissue 17.
PRECONDITIONING IN APOPTOSIS AND ISCHAEMIA–REPERFUSION LUNG INJURY
Ischaemic preconditioning achieved by brief periods of ischaemia and reperfusion before a major ischaemic insult significantly limits the degree of ischaemia–reperfusion injury to organs such as the heart, liver and kidneys. However, little is known about similar protective effects of preconditioning on the lung. Creating ischaemic preconditioning in the lung is more complex than in the other organs because of its dual blood supply and ventilation; therefore, it is more difficult to study. For example, it has been reported that blocking pulmonary blood supply during ventilation does not lead to a reduction in ATP levels 53. However, cessation of ventilation with collapsed lung may differ from anoxic ventilation, since the mechanical effects of collapse and reinflation are more important than the ischaemic challenge itself 53. In the experimental model of lung ischaemic preconditioning, markers of ischaemia–reperfusion lung injury (i.e. malondialdehyde and glutathione) and pulmonary arterial pressure were both lower in the preconditioned group after the ischaemia–reperfusion insult compared with the unpreconditioned group 54. In addition, preconditioning-induced heat shock protein synthesis in the lung also helps maintain ion transport and alveolar fluid clearance, thereby preserving gas exchange function and reducing pulmonary vascular permeability after the pulmonary ischaemic insult 55. Interestingly, experimental studies have shown that the duration and pattern of ischaemic preconditioning are also important in achieving adequate protective effects against ischaemia–reperfusion lung injury. Repetitive rather than a single period of ischaemic preconditioning can confer better lung function preservation, as measured by pulmonary compliance following ischaemia–reperfusion lung injury 53. In addition, the concepts of whole body ischaemic or thermal preconditioning have gained popularity recently 55.
Various chemicals have also been used for lung preconditioning prior to pulmonary ischaemia–reperfusion, including inhaled NO and 3-nitropropionate 56, 57. Proposed mechanisms, whereby NO provides preconditioning, include the induction of pulmonary neutrophil apoptosis, inhibiting superoxide release by neutrophils and inflammatory cytokines from alveolar macrophages 56. An inhibitor of mitochondrial complex II, 3-nitropropionate, was also able to precondition the lungs and attenuate subsequent ischaemia–reperfusion injury by inhibiting oxidative phosphorylation and oxidative stress to the cell after reperfusion 57.
CLINICAL IMPLICATIONS
Apoptosis and its upstream pathways clearly contribute to the development of lung ischaemia–reperfusion injury and some of its associated clinical manifestations. Experimental and clinical studies have confirmed two major pathways, namely the extrinsic Fas-L–caspase and the intrinsic mitochondrial pathways, through which programmed cell death occurs in the pulmonary epithelium. Numerous regulatory mediators and supplementary pathways have been implicated in the fine tuning of the apoptotic response following lung ischaemia and reperfusion. Understanding such pathways governing lung apoptosis after ischaemia–reperfusion can open new vistas for reducing the severity of acute lung injury. For instance, pharmaceutical (carbon monoxide) 20 and biomolecular interventions (Fas-L) 58, as well as genetic manipulation by adenovirus and plasmids 15, 58, have been recently proposed. These potential therapeutic strategies could help to prevent propagation of the apoptosis pathways, and, subsequently, may reduce perioperative morbidity and mortality in patients undergoing heart and lung transplantation.
SUMMARY
It is only since the early 1990s that the significant role of apoptosis and its mediators in lung ischaemia–reperfusion injury have begun to be understood. The emerging evidence suggests that the regulation of apoptosis following lung ischaemia–reperfusion is likely to be much more complex than previously envisioned. Surprisingly, the evidence for a correlation between lung apoptosis and pulmonary dysfunction resulting from lung ischaemia–reperfusion is not entirely consistent. It is possible that the intended and highly selected cellular apoptosis in the lung after ischaemia–reperfusion may not be as damaging as previously thought. Rather, cellular necrosis, with its associated inflammation, may be more injurious to the lung. Ischaemia alone appears to have only a minor effect on lung apoptosis compared with reperfusion. The full extent of the extrinsic and intrinsic apoptotic pathways, as well as their associated regulatory factors, on apoptosis in lung ischaemia–reperfusion remains to be elucidated. The role of other caspases during lung ischaemia–reperfusion needs to be further explored at the molecular level. Future research on the upstream pathways responsible for the initiation and propagation of lung apoptosis will be fundamental in devising possible therapeutic strategies for minimising lung dysfunction after lung ischaemia–reperfusion and lung transplantation.
- Received March 10, 2004.
- Accepted August 21, 2004.
- © ERS Journals Ltd
References









