





Abstract
Endothelial injury is an important prognostic factor in acute respiratory distress syndrome (ARDS). Decreased production of vascular endothelial growth factor (VEGF) in ARDS may favour vascular lesions, since VEGF promotes endothelial survival by inhibiting apoptosis.
This study sought to document low VEGF levels in lung tissue from ARDS patients, to determine whether the cause was injury to alveolar type II cells (the main pulmonary source of VEGF) and to evaluate the vascular consequences. Lung specimens were obtained by open biopsy or autopsy from 29 patients with severe ARDS (two survivors) and five controls.
As compared with controls, homogenates of lung tissue from ARDS patients contained less VEGF (median (interquartile range) ARDS 8.2 (4.7–12.2) versus controls 28.4 (9.9–47.1) ng·g−1 protein). Increased immunostaining with surfactant protein B was seen in ARDS lungs. Extensive cellular apoptosis (terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labelling staining), including endothelial and alveolar type II cells, was demonstrated, and vascular bed density (CD31 immunostaining) decreased in ARDS lungs as compared with controls. VEGF levels were negatively correlated to apoptotic endothelial cell counts.
In conclusion, decreased vascular endothelial growth factor levels in lung tissue may participate in the decrease in lung perfusion in acute respiratory distress syndrome.
The detailed morphological description of acute respiratory distress syndrome (ARDS) by Bachofen and Weibel 1 includes a decrease in lung capillary density. Conceivably, the resulting decrease in lung perfusion may lead to an increase in alveolar dead space, which has been recently shown to be a major prognostic factor during ARDS 2. The pathophysiology of vascular lesions in ARDS is a challenging issue that has received little attention 3. Recently, Hamacher et al. 4 have demonstrated that bronchoalveolar lavage fluids of ARDS patients exhibit an ex vivo pro-apopotic activity against pulmonary microvascular endothelial cells 4. Conversely, a decrease in survival factors (anti-apoptotic) of the endothelium may be an alternative hypothesis. Low levels of vascular endothelial growth factor (VEGF) have been found in the lungs of patients with early ARDS 5, 6. This VEGF decrease may contribute to the genesis of vascular injury, inasmuch as VEGF has been demonstrated to be a major survival factor for endothelium 7. VEGF (or VEGF-A) is a highly conserved, dimeric, heparin-binding glycoprotein (molecular weight 46 kDa). At least four different VEGF transcripts resulting from alternate splicing of a single gene have been identified in human cells. VEGF121 and VEGF165 are secreted in a soluble form, whereas VEGF189 and VEGF206 remain cell-surface associated or are primarily deposited in the extracellular matrix. VEGF seems to specifically affect endothelial cell growth, survival and permeability. In the lung, VEGF is expressed primarily by epithelial cells and macrophages. VEGF is also produced by and stored in both human platelets and polymorphonuclear neutrophils. The biological activity of VEGF is dependent on interaction with specific receptors (VEGF-R1, 2, and 3), which are expressed not only by endothelial cells, but also by activated macrophages and alveolar type II epithelial cells. Endothelial survival is mediated via VEGF-R2/kinase insert domain-containing receptor (KDR) 8. The aim of this study was two-fold: to determine whether the decrease in lung VEGF is related to epithelial injury, and whether it may be associated with increased endothelial apoptosis and decreased capillary density.
MATERIALS AND METHODS
Selection of acute respiratory distress syndrome and control patients
ARDS was defined as recommended by the international American–European consensus conference 9. Lung specimens from ARDS patients were obtained by open lung biopsy or autopsy at the Sainte Marguerite Hospital, Marseille, France, and frozen immediately at -80°C for subsequent examination. Open lung biopsies were performed for clinical reasons, as detailed elsewhere 10. The controls were patients undergoing thoracic surgery to treat nonsmall cell lung cancers, in whom lung tissue (normal appearance on microscopic examination) was sampled at a distance from the tumour area and processed as for the ARDS patients. The institutional review board for human studies (Sainte Marguerite Hospital, Marseille, France) approved the study protocol.
Histopathological examination
Sections that were 5 μm in thickness were cut from frozen lung tissue. All lung samples were stained (haemalum/eosin/saffron) for standard histopathological analysis in a blind fashion by a single histopathologist (F. Lange). Patients with ARDS were classified as having early or late disease, based on symptom duration at lung sampling (<5 days or ≥5 days) and on the presence of standard histological criteria for early or late ARDS 11. Presence of an inflammatory exudate and hyaline membranes in the alveoli, denudation of the basement membrane, and oedema of the alveolar wall characterised the early exudative phase. The late fibroproliferative phase was characterised by type II pneumocyte proliferation associated with myofibroblast proliferation and/or fibrosis. This classification, based on both clinical and histological criteria, was used because it is more reliable than either set of criteria used alone; acute lung injury and resulting injury of the alveolar capillary wall can occur before the criteria for ARDS are met, and histological lesions are known to be patchy, with coexistence of early-stage and late-stage features in adjacent lung regions.
Protein expression in lung tissue homogenates
Quantitative determinations of VEGF and VEGF-R2/KDR protein expressions in tissue samples were performed using ELISAs, as recommended by the manufacturer (R and D Systems, Minneapolis, MN, USA). The VEGF ELISA kit recognises both VEGF121 and VEGF165 (soluble forms). Samples were solubilised by homogenisation in lysis buffer containing 20 mM hydroxyethyl piperazine ethane sulfonic acid, 0.5 mM ethylene glycol tetra-acetic acid, 1 mM dithiothreitiol and 0.32 M sucrose (pH 7.4) for VEGF measurement, and in Tris buffer containing phenylmethylsulfonyl fluoride and 3-(3-cholaminopropyl diethylammonio)-1- propane sulfonate for VEGF-R2 measurement. Protein content was determined using the method of Bradford (Bio-Rad, Richemond, CA, USA). VEGF and VEGF-R2 protein concentrations in each sample were normalised for total protein concentration in the tissue homogenate.
Immunohistochemistry
Apoprotein surfactant protein B
Alveolar type II epithelial cells were detected using a specific rabbit polyclonal antibody directed against surfactant protein (SP)-B (Chemicon, Temecula, CA, USA), and negative controls were run with nonimmune rabbit serum (Sigma, St. Louis, MI, USA). The current authors chose to assess SP-B because this protein is more specific of type II alveolar epithelial cells than SP-A, and SP-C is known to be downregulated in animal models of acute lung injury. Tissue sections were placed in methanol at −20°C, and then incubated in hydrogen peroxide (3% in PBS) to quench endogenous peroxidase. After rinsing, nonspecific antigenic sites were blocked by incubation with 2% normal goat serum (Sigma) in PBS with 0.05% Tween 20 for 30 min at room temperature. The slides were then incubated with anti-human SP-B antibody (at a dilution of 1:1,000) or negative control serum in a moist chamber for 2 h (room temperature). Subsequently, the slides were washed in PBS with 0.05% Tween 20, and then incubated with a horseradish peroxidase-conjugated goat anti-rabbit antibody (Catalag Laboratories, Burlingame, CA, USA) and revelation substrate (diaminobenzidine (DAB) chromogen) in the dark for 7 min. Tissue sections were counterstained with haematoxylin. All tissue sections were examined using a light microscope (final magnification 400×), and the number of positive cells was counted in 10 fields taken at random. Since the degree of tissue distension can vary across lung specimens, to ensure that the surface area assessed was the same in all specimens, the same number of alveoli in each tissue sample was evaluated.
Proliferating cell nuclear antigen
Proliferating nuclei were identified by staining for proliferating cell nuclear antigen (PCNA) with monoclonal antibody PC10 (PCNA staining kit; Zymed, San Francisco, CA, USA).
Vascular endothelial growth factor and vascular endothelial growth factor receptor 2
VEGF was detected using a specific mouse monoclonal antibody (MAB293; R and D Systems), and VEGF-R2 was detected using a mouse monoclonal antibody directed against the extracellular domain of the receptor (KDR-1; Sigma). Negative controls were run with nonimmune mouse immunoglobulin (Ig)G2B (R and D Systems) and IgG1 (Sigma). Tissue sections were placed in 1% paraformaldehyde and incubated in hydrogen peroxide (3% in PBS) to quench endogenous peroxidase. After rinsing, nonspecific antigenic sites were blocked by incubation with 2% normal goat serum (Sigma) in PBS with 0.05% Tween 20 for 30 min at room temperature. The slides were incubated with anti-human VEGF antibody (at a dilution of 1:160) and anti-VEGF-R2 antibody (at a dilution of 1:800), in the presence of 1% bovine serum albumin (BSA) or negative control serum, in a moist chamber for 2 h (room temperature). Tissues were washed in PBS, treated with biotinylated goat anti-mouse Ig (Sigma) as a secondary linking antibody, and labelled with peroxidase-conjugated streptavidin (Sigma). DAB chromogen was used to visualise the antibody, and tissues were counterstained with haematoxylin.
Endothelial cell staining and apoptosis detection
Apoptosis was assessed by the terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labelling (TUNEL) method. Briefly, apoptotic fragments were detected by adding digoxigenin-labelled nucleotides with terminal deoxynucleotidyl transferase using the APOPDETECT Plus Peroxidase in situ Apoptosis Detection Kit (Qbiogen Inc., Carlsbad, CA, USA), according to the manufacturer's instructions. Nuclei of apoptotic cells appeared brown and granular. To quantify the extent of microvascular endothelial apoptosis, co-staining with an anti-human mouse monoclonal antibody to CD31 (WM-59; PharMingen, San Diego, CA, USA) was used, which recognises the endothelial surface marker platelet/endothelial cell adhesion molecule 1. TUNEL sections were incubated with 2% normal goat serum (in PBS with 0.05% Tween 20), and, subsequently, with the primary anti-CD31 antibody (1:100,000 dilution, in PBS with 1% BSA) overnight at 4°C. Mouse nonimmune IgG1 (Sigma) was used as the negative control. Biotinylated goat anti-mouse antibodies (Sigma) were applied for 30 min at room temperature, followed by avidin-biotin peroxidase (Sigma) complexes for 30 min (room temperature). True Blue Peroxidase substrate was used as the final chromogen (KPL Laboratories, Gaithersburg, MD, USA). No counterstaining was used, to avoid possible interference with the specific dark-blue immunostaining of the endothelial cell surface. Double staining was considered positive when specific cells displayed a brown nucleus with a surrounding blue-to-black membrane-immunoreactive pattern. Positive cells were counted in 10 randomly selected areas (final magnification 400×).
Image analysis
A charge-coupled device Iris camera (CDD Iris; Sony France, Paris, France), coupled with an optical microscope (Leitz Laborlux, Wetzlar, Germany), was used to view the sections and to digitise colour images to a PC host computer. CD31 and TUNEL staining were estimated with Perfect Image software (ClaraVision, Orsay, France), which differentiates colours based on their red–green–blue proportions with a ±10% variation. At least 10 fields per section were analysed, with approximately the same number of alveoli. Results are given as the ratio (%) of the blue- (CD31) or brown- (TUNEL) stained surface area over the total field surface area (CD31 or TUNEL staining relative to total field surface).
Statistical analysis
Results are presented as medians (interquartile range) in the text. Data are presented as box-plots indicating the median and the 10th, 25th, 75th and 90th percentiles. Differences between groups were estimated using the Kruskal-Wallis test with post hoc Mann-Whitney analysis. To assess the possible influence of VEGF on pulmonary microcirculation, the relationships linking VEGF levels to endothelial quantification and apoptosis were assessed by Spearman's rank correlation. p-Values <0.05 were considered statistically significant.
RESULTS
Patient characteristics
A total of 29 ARDS patients were studied (20 males and nine females; median age 64 yrs (50–73)). Their median acute physiology and chronic health evaluation II score at admission was 16.9 (13.6–23.6). Open lung biopsy was performed as a diagnostic procedure in 15 patients and immediately after death in 14 patients.
At the time of lung tissue sampling, the arterial oxygen tension/inspiratory oxygen fraction ratio was <200 mmHg in 22 patients and 200–300 mmHg in seven patients. The cause of ARDS was direct lung injury in 17 patients (related to infection in 11 and to inhalation in six) and indirect lung injury in 12 patients (nonpulmonary sepsis in seven, nonseptic shock in four, and pancreatitis in one). The six patients who died within 5 days after ARDS onset were classified as having acute ARDS, whereas the remaining 23 patients were classified as having late ARDS; histological findings confirmed the classification in every case. Survival was only 7% (two out of 29) in this selected population.
The control group was composed of five patients (all males; median age 65 years (54–70)).
Vascular endothelial growth factor expression in lungs from acute respiratory distress syndrome and control patients
VEGF levels in the lung homogenates were lower in the ARDS patients than in the controls: 8.2 (4.7–12.2) versus 28.4 (9.9–47.1) ng·g−1 protein, respectively (p = 0.03). There was no difference between the early and late ARDS subgroups (fig. 1⇓). Results were similar when VEGF concentrations were normalised per gram of lung weight. In the controls, immunostaining for VEGF (fig. 2⇓) labelled the bronchiolar cells and alveolar macrophages and strongly labelled some alveolar type II epithelial cells. Alveolar type I epithelial cells were negative. In the ARDS patients, staining was highly heterogeneous. Bronchiolar and alveolar epithelial cells were negative in some areas and positive in others, even within a tissue section. In early ARDS tissue samples, inflammatory cells were also positive.

Vascular endothelial growth factor (VEGF) concentrations in the lung homogenates of patients with early-stage (<5 days; n = 6) or late-stage (≥5 days; n = 23) acute respiratory distress syndrome (ARDS) and of control patients (n = 5). VEGF was measured by immunoassay. The box-plot shows the median and the 10th, 25th, 75th and 90th percentiles. *: p<0.05 compared with control patients.
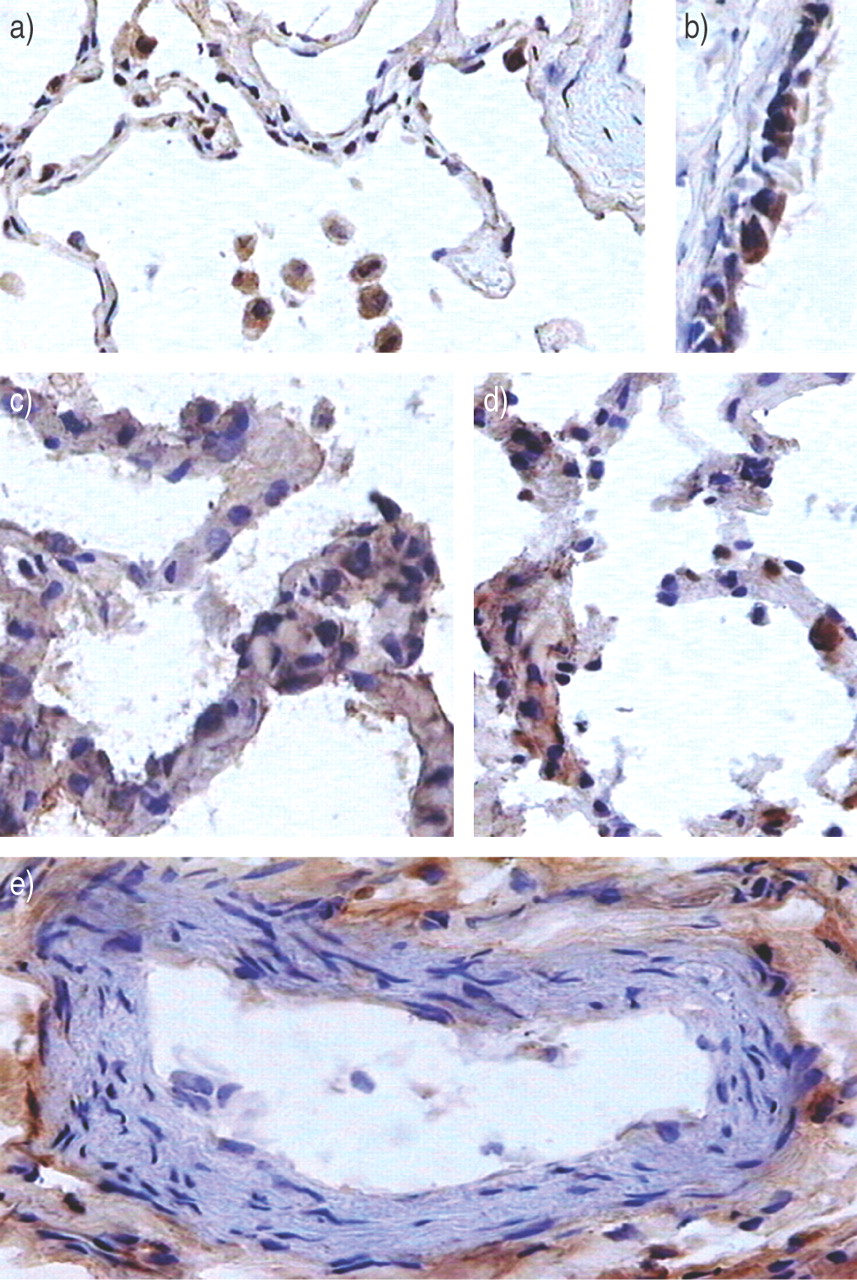
Representative immunoperoxidase staining for vascular endothelial growth factor (brown staining). a) In control patients, immunostaining was seen in some alveolar type II epithelial cells and in some alveolar macrophages. b) Airway epithelial cells were positive. c, d) In patients with late acute respiratory distress syndrome, the alveolar capillary wall was clearly modified; immunostaining was heterogeneous. Staining seemed to be extracellular in some areas, such as in perivascular spaces (e).
Vascular endothelial growth factor receptor 2 expression in lungs from acute respiratory distress syndrome and control patients
There was no significant difference between VEGF-R2 concentrations measured by ELISA in lung homogenates from ARDS and control patients (116 (78–157) versus 156 (131–191) ng·g−1 protein). No difference was found between early and late ARDS patients.
Immunostaining for VEGF-R2 was noted in endothelial cells in the control group; however, not all endothelial cells were positive. Macrophages, epithelial cells and bronchiolar cells were negative. In ARDS groups, staining extended to some alveolar type II epithelial cells (data not shown).
Alveolar epithelial type II cell proliferation in acute respiratory distress syndrome patients
Proliferation of alveolar epithelial type II cells was found in the ARDS patients as compared with the controls: 13 (11–17) versus 7 (5–8) cells per field, respectively (p = 0.003; fig. 3⇓). Furthermore, the number of alveolar epithelial type II cells was greater in the patients with late than with early ARDS: 16 (12–17) versus 11 (8–12) cells per field, respectively (p = 0.02; fig. 4⇓). Despite an increased number of alveolar epithelial type II cells in ARDS patients, PCNA staining was rare.

Quantification of the number of alveolar type II epithelial cells per optical field (surfactant protein B staining) in lung biopsies of patients with early-stage (<5 days; n = 6) or late-stage (≥5 days; n = 23) acute respiratory distress syndrome (ARDS) and of control patients (n = 5). The box-plot shows the median and the 10th, 25th, 75th and 90th percentiles. *: p<0.05; #: p<0.02; ¶: p<0.005.

Representative immunoperoxidase staining for surfactant protein B in a control patient (a) and acute respiratory distress syndrome patients with alveolar type II epithelial cell proliferation covering the alveolar barrier leading to an increased alveolar capillary wall thickness (b, c, and d). The cytoplasm of alveolar type II epithelial cells is stained brown.
Decreased pulmonary endothelial density in acute respiratory distress syndrome patients
Endothelial density was estimated using computer-assisted quantification of CD31 staining (figs 5⇓ and 6⇓). Endothelial area was significantly lower in the ARDS patients than in the controls: 3.5% (2.3–4.1) versus 5.3% (4.7–6.7) of the total field area, respectively (p = 0.01). In contrast, no difference was found between early and late ARDS. In more than two-thirds of the ARDS patients, marked heterogeneity was seen in the distribution of the pulmonary endothelium.
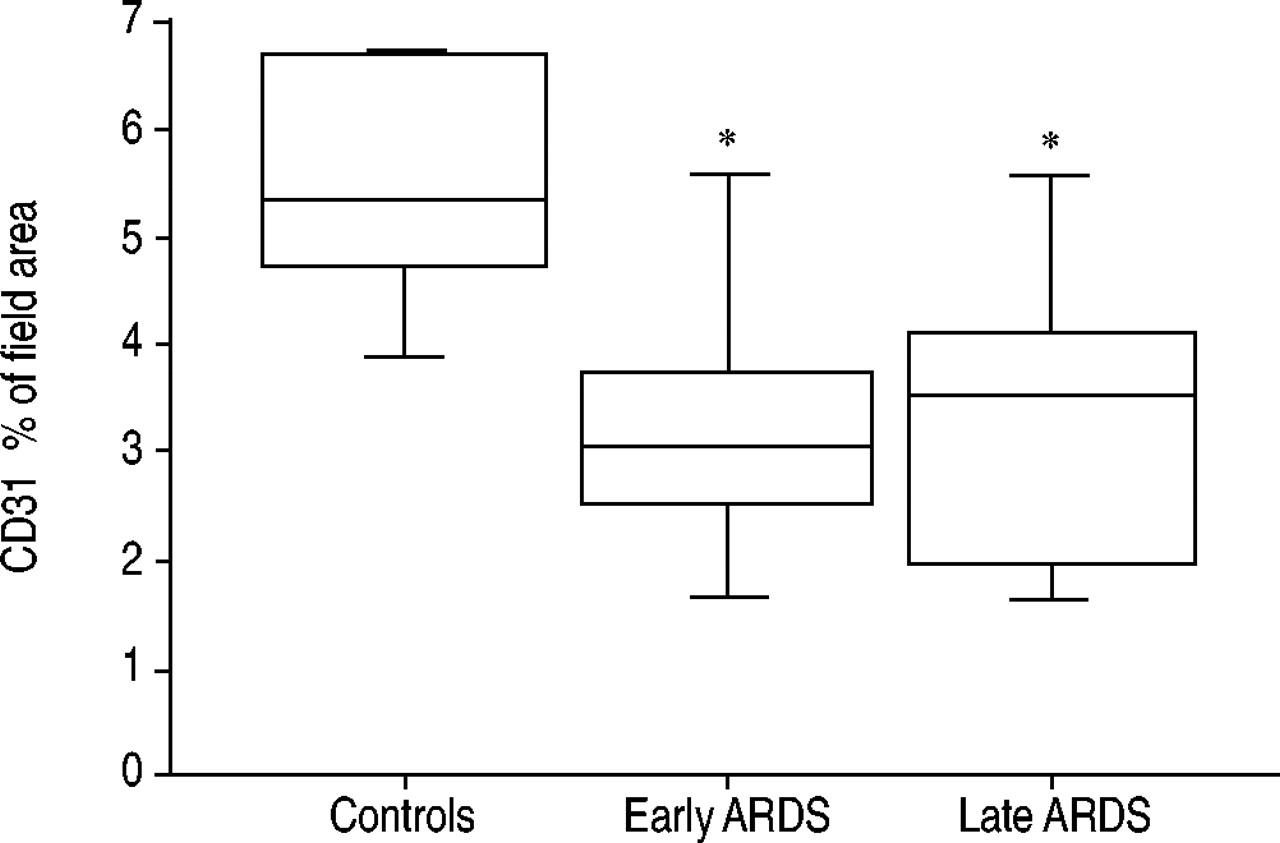
Automatic quantification of endothelial staining (image analysis; see Methods) of lung biopsies from patients with early-stage (<5 days; n = 6) or late-stage (≥5 days; n = 23) acute respiratory distress syndrome (ARDS) and from controls (n = 5). Results are given as a percentage of the CD31 stained area over the total field area. The box-plot shows the median and the 10th, 25th, 75th and 90th percentiles. *: p<0.05 compared with control patients.

Endothelial evaluation using double immunostaining for terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labelling/CD31 in lung tissue from a control patient (a) and a patient with acute respiratory distress syndrome with a moderate decrease in vascular bed density (b). The endothelial surface is stained blue-to-dark blue with True Blue Peroxidase substrate (KPL Laboratories, Gaithersburg, MD, USA) as the final chromogen. No counterstaining was used.
Increased endothelial cell apoptosis in acute respiratory distress syndrome patients
The number of apoptotic endothelial cells was greater in the ARDS patients than in the controls, as shown by the number of cells double-labelled for TUNEL and CD31: 8 (6–13) versus 4 (3–4) cells per 10 optical fields, respectively (p = 0.005; fig. 7⇓). No significant difference was found between early and late ARDS, or between open lung biopsy and autopsy specimens from ARDS patients. A negative correlation was observed between pulmonary VEGF concentrations and the number of apoptotic endothelial cells (ρ = −0.40; p = 0.02).

Evaluation of the number of apoptotic endothelial cells per 10 optical fields. This count was performed using terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labelling/CD31 co-staining in lung sections from patients with early-stage (<5 days; n = 6) or late-stage (≥5 days; n = 23) acute respiratory distress syndrome (ARDS) and from controls (n = 5). The box-plot shows the median and the 10th, 25th, 75th and 90th percentiles. *: p<0.05 compared with control patients; #: p = 0.004 compared with control patients.
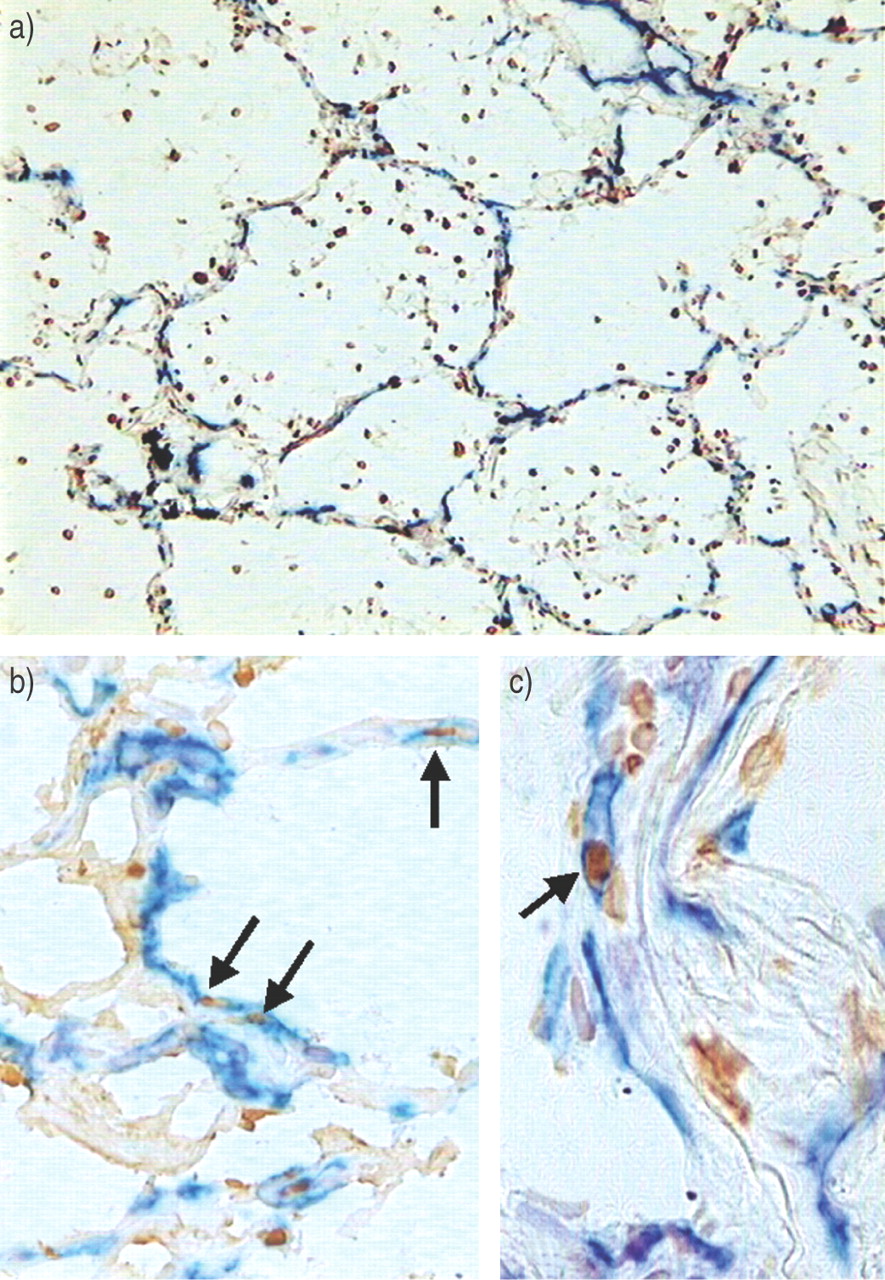
An additional result from TUNEL staining was to demonstrate extensive apoptosis in the lungs of ARDS patients as compared with controls: 4.0% (3.0–4.0) of surface area versus 2.0% (1.0–3.2), respectively (p = 0.03). Increased apoptosis involved endothelial cells and particularly inflammatory cells and alveolar epithelial type II cells (fig. 8⇓).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Representative double immunostaining is shown for terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labelling (TUNEL)/CD31, revealing extensive apoptosis involving alveolar inflammatory cells (a), epithelial cells (b) and endothelial cells (c) in lungs from patients with acute respiratory distress syndrome. Apoptotic endothelial cells (arrows) have brown nuclei (TUNEL) and a blue-to-black membrane (CD31). A severe loss of vascular bed is shown in this patient (b).
DISCUSSION
This study confirms that pulmonary VEGF levels are low early in the course of ARDS, and suggests that this decrease may contribute to the increase in endothelial apoptosis and, consequently, to shrink the pulmonary vascular bed.
The decrease in lung VEGF levels was expected, since two earlier studies documented an early decrease in VEGF concentrations in epithelial lining fluid from ARDS patients 5, 6. Inasmuch as this decrease in VEGF concentration in epithelial lining fluid raised several concerns, it was useful to confirm this result in lung tissue. A limitation of the current study is that control patients were suffering from malignant tumours, even if tissue sampling was performed at distance. However, upregulation of VEGF is not a constant finding in nonsmall cell lung cancers 12, and increased VEGF expression is associated with areas of increased microvessel density 12, which was not evidenced in the current control patients. A decrease in lung VEGF has also been described in other forms of lung injury in humans, including respiratory distress syndrome in infants, pulmonary bronchodysplasia and idiopathic fibrosis 13–15. Inasmuch as several factors may affect lung VEGF levels in subjects, including smoking effects and underlying chronic lung diseases, it cannot be ensured that the low levels of VEGF concentration were ascribable to ARDS in this study. However, VEGF concentration in epithelial lining fluid of patients with ARDS increased with time in survivors, and the persistence of low levels of VEGF were associated with lower survival rates in the study by Thickett et al. 6. In the current study, VEGF levels were low in both early and late ARDS. The extremely high mortality rate in the current patients (∼93%) suggests that low concentrations of VEGF, whatever the stage, may be ascribable to this selection bias. The degree of vascular injury has been shown to be an important prognostic factor, in keeping with earlier evidence of decreased survival in ARDS patients with higher alveolar levels of von Willebrand factor (a marker for endothelial injury), more severe pulmonary hypertension or a larger dead space 2, 11, 16. Interestingly, acute endothelial injury has been reported to be more pronounced at the proliferative phase than at the exudative phase of ARDS 11. Although VEGF levels are low in the bronchoalveolar lavage fluids of ARDS patients, an increase in angiogenic activity related mainly to CXC chemokines has been reported in one study 17.
Despite the documentation of an early (fourth hour) decrease in VEGF mRNA in an acute lung injury model in rats 5, these findings do not rule out an increase in VEGF release at the early onset of ARDS, from neutrophil granules undergoing exocytosis during neutrophil migration and from platelets aggregated in the vascular bed. This could explain the increase in VEGF in plasma that has been confirmed during ARDS 18 and the apparent increased extracellular VEGF immunostaining in some areas in this study. Thus, a role for VEGF in the initial increase in endothelial permeability that characterises ARDS cannot be excluded. In keeping with this possibility, adenoviral-induced overexpression of VEGF in healthy animals is associated with increased oedema formation 19. Interestingly, in the present study, VEGF immunolocalisation labelled alveolar inflammatory cells, in agreement with the finding by Thickett et al. 6, which demonstrated that VEGF was released by isolated inflammatory cells.
A limitation of this study is that the current authors did not determine which VEGF isoforms were decreased. However, in a previous experimental study, it was demonstrated that bacteria-induced lung injury was associated with decreases in mRNAs for VEGF121, VEGF165 and VEGF189 5. Recently, Tsokos et al. 20 have demonstrated a decrease in VEGF protein (using ELISA and immunohistochemistry) in the lung tissue of critically ill patients dying from sepsis, as compared with nonseptic control individuals, associated with a decrease in mRNAs for VEGF121 and VEGF165.
It has been previously argued that the VEGF decrease associated with ARDS was probably not a result of damage to alveolar type II epithelial cells, inasmuch as the main function of these cells, i.e. fluid clearance, was preserved in a bacteria-induced model of acute lung injury 21 in which decreases in both VEGF mRNA and VEGF protein were shown 5. The number of alveolar type II epithelial cells was increased, in keeping with previous studies in ARDS demonstrating similar increases in numbers, as well as elevation of epithelial growth factors 1, 22, 23. It was not demonstrated why VEGF release diminishes in the lung, and the current authors have previously hypothesised that, during the post-injury proliferative phase, epithelial cells do not secrete VEGF 5. From the current results, it may be hypothesised that alveolar epithelial type II cells undergoing apoptosis do not secrete VEGF. Only one assay (TUNEL) was used to assess apoptosis; consequently, a clear distinction with necrosis cannot be made. However, several experimental studies have demonstrated a component of apoptosis during the resolution of type II cell hyperplasia following lung injury 24. Bardales et al. 25 demonstrated that apoptosis is a major pathway responsible for the resolution of alveolar type II epithelial cells in ARDS patients. In this latter study, the authors demonstrated that most patients exhibited type II pneumocytes apoptosis (30–80%), together with extremely rare PCNA positivity 25, which is in agreement with the current results. In the study of Albertine et al. 26, all antigens for proapoptotic markers were increased in patients with ARDS as compared with controls, and they further suggested that markers of apoptosis were not likely to be influenced by the use of autopsy samples.
The causes or consequences of the decrease in lung VEGF were indirectly assessed in this study, inasmuch as a decrease in VEGF could induce or be related to microvessel density decrease. The expression of the VEGF receptor was evaluated, which is known to mediate the anti-apoptotic effect. VEGF-R2 expression was not different between ARDS patients and controls, which could seem contradictory to the decreased vascular bed. This could be explained by the fact that VEGF-R2 expression is not restricted to endothelial cells, since alveolar type II epithelial cells express VEGF-R2 27. The number of apoptotic endothelial cells was greater in the ARDS patients. A negative correlation was found between VEGF levels and the number of apoptotic endothelial cells, further emphasising the possible role of VEGF on endothelial cell survival. It has to be noted that this statistical correlation does not imply a causality relationship. VEGF is probably not the only factor involved in endothelial cell apoptosis. At least three studies found increased levels of pro-apoptotic soluble factors in bronchoalveolar lavage fluids from ARDS patients 4, 26, 28. Endothelial cell apoptosis seems to lead to shrinkage of the pulmonary vascular bed. Results from animal models of sepsis support this possibility: systemic endotoxin administration induces both a decrease in lung VEGF and apoptosis of endothelial cells 29, 30. Consequently, improving lung endothelial survival may produce clinical benefits in patients with ARDS. Administration of a caspase inhibitor has been shown to prevent lethal apoptosis in mice with endotoxin-induced acute lung injury 31. Compernolle et al. 32 demonstrated a beneficial effect of VEGF administration during respiratory distress syndrome in neonatal mice 32, and Corne et al. 33 found that mice transfected with interleukin 13 during hyperoxia-induced lung injury had an increase in survival that was mediated in part by an increase in VEGF synthesis 33.
CONCLUSIONS
Lung vascular endothelial growth factor levels were low in this selected population of acute respiratory distress syndrome patients with a very high mortality rate. An increase in endothelial cell apoptosis and a decrease in capillary density were found. Further studies are warranted to determine whether inhibition of endothelial apoptosis may provide clinical benefits in patients with acute respiratory distress syndrome.
- Received June 4, 2004.
- Accepted September 20, 2004.
- © ERS Journals Ltd
References









