





Abstract
Primary pulmonary lymphangiectasia (PPL) is a rare disorder of unknown aetiology characterised by dilatation of the pulmonary lymphatics. PPL is widely reported to have a poor prognosis in the neonatal period and little is known about the clinical features of patients who survive the newborn period.
The current authors report the outcome in nine patients diagnosed in infancy with PPL over a 15-yr period at a single university-based hospital clinic and followed for a median of 6 yrs.
Although all of the patients initially experienced respiratory distress, respiratory symptoms improved in most patients after infancy and were notably better by the age of 6 yrs. Many patients had poor weight gain in the first years of life, which eventually improved. Radiological scans showed progressive resolution of neonatal infiltrates, but were characterised by hyperinflation and increased interstitial markings in older children. Most patients had evidence of bronchitis and grew pathogenic organisms from quantitative bronchoalveolar lavage culture. Pulmonary function tests showed predominantly obstructive disease that did not deteriorate over time.
In conclusion, these results suggest that primary pulmonary lymphangiectasia does not have as dismal a prognosis as previously described and symptoms and clinical findings improve after the first year of life.
C.R. Esther Jr is supported by a Parker B. Francis Pulmonary Fellowship.
Pulmonary lymphangiectasia is an abnormal dilatation ofthe lymphatics draining the interstitial and subpleural (SP) space of the lungs. It is a rare condition, first described by Virchow 1 in 1856, which can be a primary abnormality or acquired as a result of obstruction of the pulmonary lymphatics or veins. In primary pulmonary lymphangiectasia (PPL), the lymphatic abnormality can belocalised to the lung or be part of a more widespread abnormality of lymphatic drainage 2. Although PPL is assumed to result from an arrest in the development of thepulmonary lymphatics 3, the cause of PPL is not known. Most cases are sporadic and, although familial occurrences ofthis condition have been described 4–6, no genetic aetiology has been identified. PPL has been described in association with a number of chromosomal anomalies 7 andin patients with Noonan's syndrome 8–11. In addition, PPL is often described in association with cardiac anomalies 12 that are not associated with pulmonary venous hypertension.
Early descriptions of pulmonary lymphangiectasia from the pathology literature reported an incidence of ∼1% in consecutive necropsies on stillbirths and neonates 12, 13. The true incidence of PPL after the newborn period is not known and reports of PPL in infants and children are restricted to isolated case reports. Despite a number of case reports in which infants with PPL have survived and improved over time 2, 5, 11, 14–17, congenital pulmonary lymphangiectasia continues to be considered as a condition with a poor prognosis 15, 18.
The current study describes the presentation, clinical findings, diagnosis, clinical course and outcome of infants diagnosed with PPL.
Methods
Study subjects and design
Retrospective chart review was used to identify nine patients who were diagnosed with pulmonary lymphangiectasia in a university hospital-based paediatric pulmonology practice (North Carolina Children's Hospital, University ofNorth Carolina, Chapel Hill, NC, USA) over a 15-yr period (1987–2002). Evaluation of patients with PPL (n=9) included: radiological studies with chest radiograph (n=9) andchest computed tomography (CT) scan (n=5); flexible fibreoptic bronchoscopy (n=8); sweat chloride test (n=8); cardiac evaluation with echocardiography or catheterisation (n=8); pH probe for gastroesophageal reflux (n=7); andan immunological evaluation, including complete bloodcount with differential and immunoglobulin levels(n=8).
To determine the frequency of a diagnosis of PPL in infants and children who underwent open lung biopsy, the current authors reviewed the pathology reports of all open lung biopsies in infants and children over a 3-yr period (1999–2002).
Results
Patients
Nine patients were identified with a diagnosis of PPL from ∼17,000 new patients seen in the current authors' clinic (North Carolina Children's Hospital) during the period under review (table 1⇓). Eight of the nine patients were males. The patients were followed for a median of 6 yrs (range 3 months to 14 yrs) and ranged between 22 months and 17 yrs of age at the time of the current study. Two patients were lost to follow-up when they relocated to another area; another patient died of respiratory disease at 3 months of age.
Characteristics of patients with primary pulmonary lymphangiectasia
Presentation
The median age at presentation was 3 months (range: newborn–12 months; table 1⇑). Three of these patients presented in the neonatal period; the remaining six patients presented in the post-neonatal period. The diagnosis of PPL was made on the basis of lung biopsy in eight patients and classic clinical presentation, including respiratory distress and chylothorax, in one patient (patient 1). Three patients had some degree of generalised oedema at presentation, which resolved, but none of these patients had intestinal lymphangiectasia, which characterises the general form of lymphangiectasia as described by Noonan et al. 2.
Three of the patients required mechanical ventilation, all of whom presented in the neonatal period. One infant with generalised oedema had severe respiratory failure and was considered for extracorporeal membrane oxygenation. None of the patients who presented in the postneonatal period required mechanical ventilation, but three required home supplemental oxygen for some period of time. All but one of these patients had fine crackles on lung auscultation with clinical evidence of decreased lung compliance (intercostal retractions, increased work of breathing) at presentation. Wheezing was present in some cases and was variably responsive to albuterol. Respiratory symptoms improved in all patients who survived the neonatal period. The patient who died had Costello's syndrome and support was withdrawn at the age of 3 months.
Other diagnoses were common in these patients. One patient was diagnosed with Noonan's syndrome and had echocardiographic evidence of mild pulmonary valvular stenosis. The patient with Costello's syndrome also had mild pulmonary valvular stenosis. None of the other patients had clinically apparent chromosomal abnormalities. All but one of the patients had echocardiogram or cardiac catheterisation and no other major cardiac anomalies were detected. One patient did have pulmonary hypertension and tricuspid regurgitation noted on the first day of life, but this was resolved later. Gastroesophageal reflux disease was present in four patients and decreased immunoglobulins were detected in two patients.
Growth
Growth data up to the age of 3 yrs was available for six of the patients in this study and weight percentiles were derived from the Center for Disease Control 2000 population growth data for males aged 0–36 months 19. All of these patients were born at term with normal birth weights, but showed poor weight gain and some degree of growth failure between the ages of 3 months and 1 yr (fig. 1⇓). By 3 yrs old, four of the six had regained normal growth. Two other patients did not have evidence of growth failure and another died at three months old.

Growth of patients with primary pulmonary lymphangiectasia. Weights at precise 3 and 6-month intervals were determined by linear extrapolation from growth data for five male patients between birth and 36 months. Weight percentiles are derived from Center for Disease Control 2000 population growth data for males aged 0–36 months 19. –––: average and standard deviation of patient weights; ····: 5th and 95th percentiles of population weight; ----: 50th percentile of population weight.
Lung biopsy
PPL was confirmed in eight of the nine patients with open lung biopsy. No complications occurred in any patients during surgery. In one case, the diagnosis of pulmonary lymphangiectasia was not made until the pathology was re-reviewed 3 yrs after the original biopsy. In two other cases, the pulmonary lymphangiectasia was not evident on the first lung biopsy. In these two cases, initial lung biopsy showed diffuse alveolar damage or nonspecific changes with no evidence of lymphatic dilation. The patient who did not have a lung biopsy had generalised lymphoedema and chylothorax at birth. Lung biopsy was deferred because the patient's clinical condition improved. For the purposes of the current study, all available lung biopsies (n=6) from these patients were reviewed independently by two anatomic pathologists for lymphatic dilation in SP, peri-arterial (PA) and interlobular (IL) regions. In these patients, lymphangiectasia involved all of the regions that were examined (fig. 2⇓). The lymphatic dilation was described as “severe” in five of six re-reviewed cases and “moderate” in one of six.

Lung pathology in primary pulmonary lymphangiectasia. These are representative images of one of the patients in the study, demonstrating lymphatic dilation in three contiguous lymphatic sites: peri-arterial lymphatic dilation (a and b); subpleural lymphatic dilation (c and d); and interlobular septal lymphatic dilation (e and f).
To exclude the possibility of lymphatic dilation that may be an artefact and associated with the cross-clamp wedge biopsy technique, control groups of all paediatric open lung biopsies over a 3 yr period (1999–2002) and all adult wedge biopsies from 2002 were reviewed. A total of 42 wedge biopsies in 31paediatric patients were evaluated. Focal lymphatic dilation was detected in six of 42 biopsies (14%) from four of 31 patients (13%). Two patients had pulmonary lymphangiectasia in a single region: one with osteosarcoma and extensive lung metastases (SP dilation in two of four biopsies) and another patient evaluated at 14 months for persistent tachypnoea (SP dilation in two of two biopsies). One patient with bronchopulmonary dysplasia and persistent tachypnoea had lymphatic dilation in two regions (SP and IL) of a single-wedge biopsy at 3 months old. One patient with known pulmonary veno-occlusive disease had lymphatic dilation in all three regions in a single-wedge biopsy obtained for the evaluation of a persistent radiological density. In the adult control group, 48 biopsies from 38 patients were reviewed. Single-site lymphatic dilation (SP: n=4; IL: n=2; PA: n=2) was observed in seven of 48 biopsies (14%) from seven of 38 patients (18%). These seven biopsies demonstrated carcinoma (n=4) and interstitial lung disease (n=3).
In summary, PPL biopsies showed lymphatic dilation involving three contiguous sites (SP, IL and PA) in all wedge biopsies, whereas control paediatric and adult wedge biopsies performed for non-lymphangiectasia lung diseases typically showed, at most, single-site lymphatic dilation and, then, in only 15% of biopsies.
Radiology
Radiological findings for four of these patients have been described previously 20. Two of the patients in the current authors' study had pleural effusions (figs 3a and b⇓). Initially, the three patients with a neonatal presentation had diffuse interstitial infiltrates. With the resolution of clinical symptoms, chest radiographs became progressively clearer, but with the emergence of increased interstitial markings and hyperinflation (figs 3c and d⇓). Patients who presented outside the neonatal period had hyperinflation and increased interstitial markings at presentation, which improved over time (figs 4a and b⇓). CT scans showed considerable parenchymal inhomogeneity with patchy areas of ground-glass infiltrates in the perihilar and SP regions. In two patients, lung disease was monitored by repeated CT scans (over an 18 month and 5 yr period, respectively). Although there was interval improvement in both patients, the pattern of parenchymal inhomogeneity was observed to persist in patient-specific regions of the lung (figs 4c and d⇓).

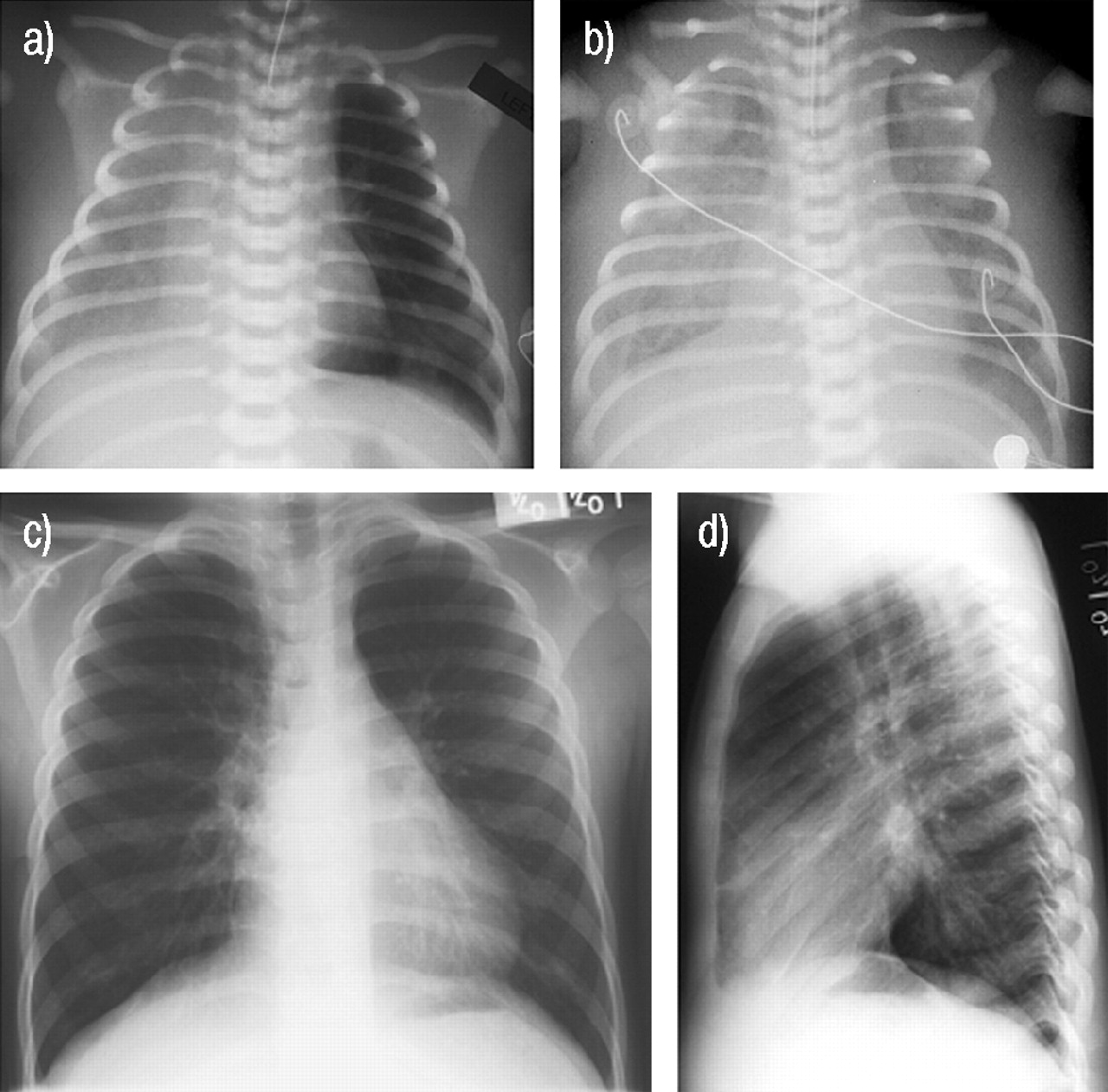
a) Radiograph from the first day of life demonstrates bilateral pleural effusions, with the right much greater than the left, a normal sized heart and relatively normal lung parenchyma with some interstitial prominence. An endotracheal tube is in place. b) A radiograph 1 week later reveals bilateral pleural effusions, with the right greater than left, and mild bilateral interstitial prominence with an endotracheal tube in place. There is also significant body wall oedema. c, d) Follow-up radiographs 5 yrs later demonstrate mild hyperinflation and minimal interstitial prominence.
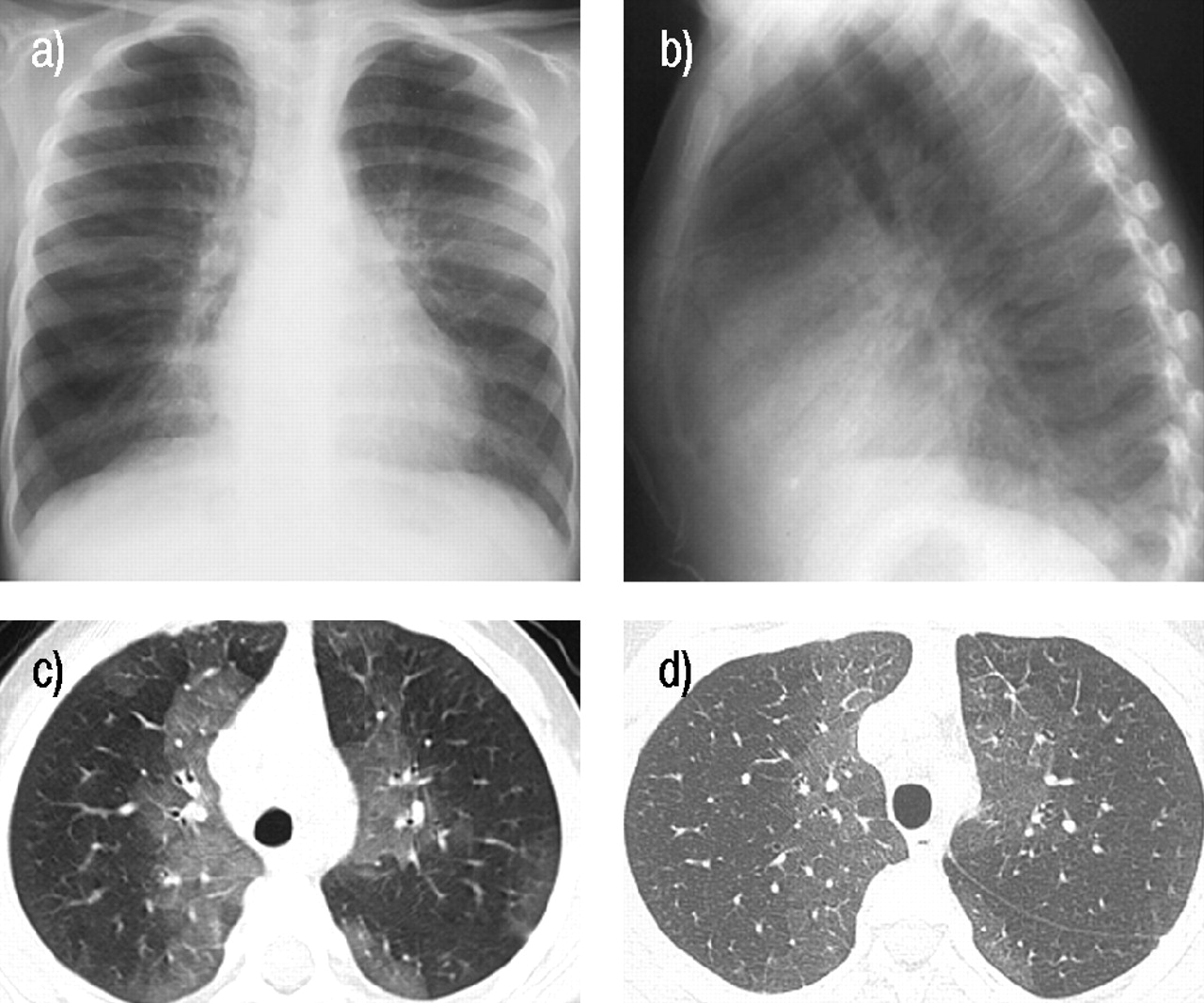
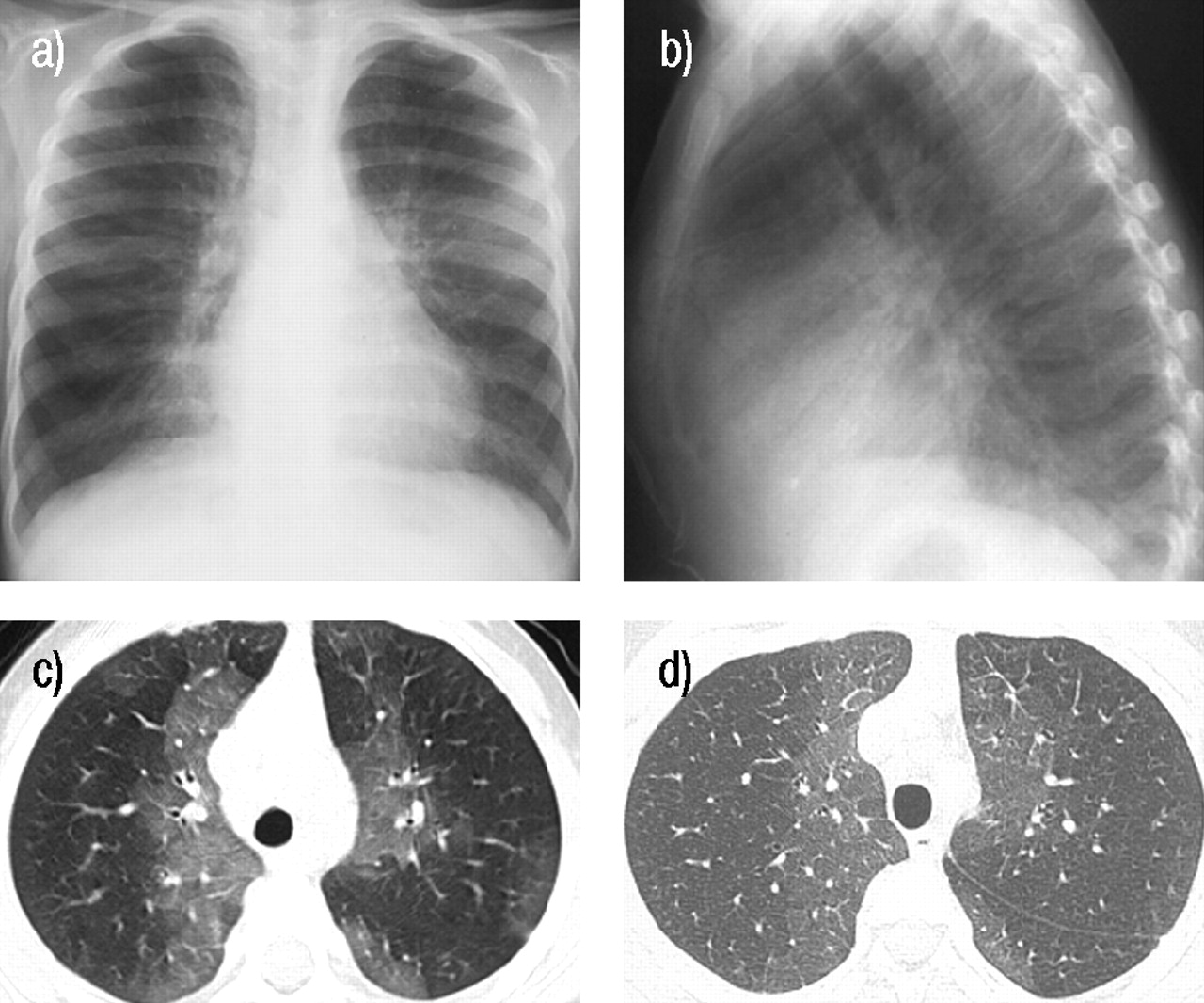
a) Anterior-posterior chest radiograph at aged 4 yrs reveals perihilar and peripheral patchy opacity and hyperinflation. b) Lateral chest radiograph from the same time as a) demonstrates a mild pectus excavatum configuration to the anterior chest wall. c) High resolution computed tomography (CT) scan from the same period demonstrates perihilar and peripheral ground-glass opacities. d) Repeated high resolution CT scan 5 yrs later at aged 9 yrs demonstrates similar findings at the same level with a slight decrease in the ground-glass component.
Pulmonary function tests
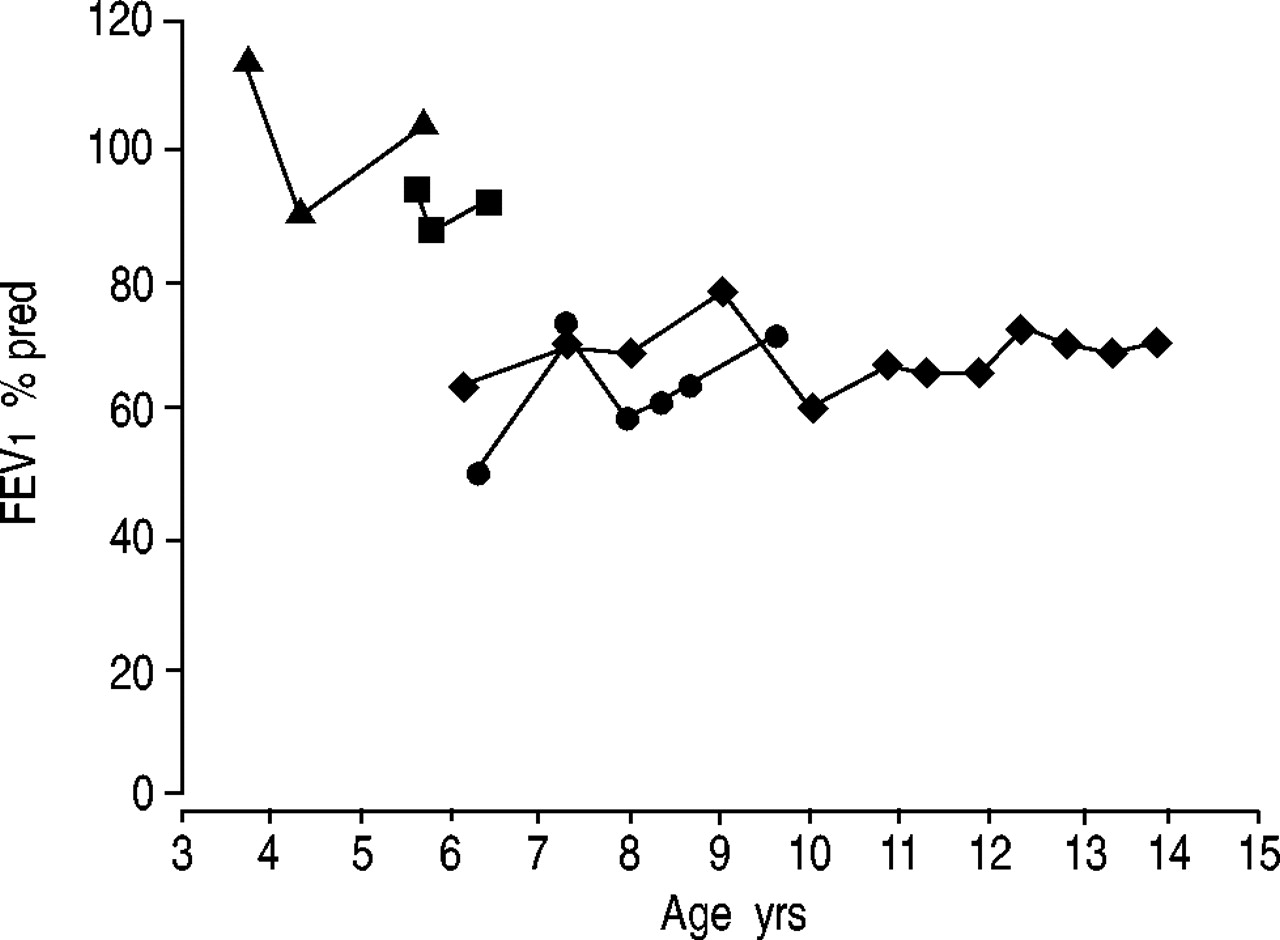
Spirometry was performed on six of the nine patients (patients 1, 3, 4, 5, 7 and 8). Of the three missing patients, one patient died in infancy and the remaining two survivors were not old enough for reliable pulmonary function testing (PFT) at the time of the current study. Spirometry values obtained at the age of 6 yrs were compared. Four of the six patients had decreased forced vital capacity (FVC) and forced expiratory volume in one second (FEV1) with a relatively normal FEV1/FVC ratio. Two patients had small airways obstruction which improved after bronchodilator treatment, including one of thepatients with decreased FVC. One patient had normal spirometry. Lung volume measurements were obtained from three patients aged 7–14 yrs, all of whom had decreased FVC and FEV1 at the time of the measurement. These studies revealed relatively normal total lung capacity (range: 81–117% predicted) with increased residual volume (range: 139–234% pred) suggesting obstructive lung disease. Diffusion capacity obtained from two patients was normal. Longitudinal PFT data was available for four of the remaining seven patients. In four patients, in whom lung function was tracked by spirometry, FEV1 did not change significantly over time (fig. 5⇓).

Spirometry over time in patients with primary pulmonary lymphangiectasia. Spirometry data was obtained at each clinic visit according to American Thoracic Society guidelines. Forced expiratory volume in one second (FEV1) over time is shown in the four patients (▴, ▪, ♦ and •) in whom reliable spirometry data could be obtained. There is no apparent decrease in FEV1 over time in these patients.
Bronchoscopy
Eight of the nine patients underwent elective flexible fibreoptic bronchoscopy as part of a diagnostic evaluation for chronic lung disease. Three patients with recurrent lung infections underwent multiple procedures to assist with the identification of pathogenic organisms. Airway anatomy was normal in six of the eight patients; mild subglottic stenosis was noted in two. Bronchitis (increased secretions and neutrophil percentage on bronchoalveolar lavage fluid) was apparent on at least one occasion in six of the eight patients studied. In seven of the eight patients, bronchoalveolar lavage cultures grew pathogenic organisms including Moraxella catharralis and Staphylococcus aureus. In three of these patients, significant quantities (>100,000 organisms·mL−1 bronchoalveolar lavage fluid) of organisms were isolated.
Discussion
The cases described in the current study suggest that PPL has a variable presentation and the widespread characterisation of this condition as having a near-uniform poor prognosis needs to be revised. Although most patients had tachypnoea (increased respiratory effort with inspiratory crackles and failure to thrive), these symptoms improved over time.
Since this is a retrospective review of cases from one tertiary institution, the current authors were unable to accurately determine the incidence of PPL over this period. Although no neonatal death was ascribed to PPL during this period, it is possible that the diagnosis was not recognised in some cases of severe neonatal respiratory failure that resulted in early death. In addition, it is possible that a number of patients with mild disease were not diagnosed in infancy because of spontaneous regression of their disease.
The studied patients fell into two broad categories: those with neonatal or postneonatal onset. In the three patients with neonatal onset, all required prolonged mechanical ventilation, one required chest tube placement for removal oflarge amounts of chylous effusion and another had persistent unilateral nonchylous effusion. In the current study, respiratory symptoms improved over time in most patients, even in those who presented in the neonatal period. This contrasts with a study of 11 patients with PPL reported recently by Bouchard et al. 15 in which none of the six patients diagnosed in the neonatal period survived. However, two of these infants were <30 weeks gestation and the remaining four had complex congenital cardiac anomalies. As such, it is difficult to know the extent of contribution of PPL to the outcome in these infants.
Many of the previous reports that built the consensus about the poor outcome for PPL come from an era of limited neonatal intensive care capabilities and/or included infants with significant additional congenital or chromosomal abnormalities 3, 6, 12, 21. Whilst there are undoubtedly infants with extensive lymphangial derangements that may not be compatible with life, it is possible that many of the infants reported previously with uncomplicated PPL would have survived with current practices of neonatal intensive care. This assertion is supported by a recent study of an infant with respiratory failure and severe chylous drainage who survived following aggressive neonatal respiratory support 22. The current study suggests that survival of neonatal onset PPL is not uncommon and that, in survivors, the condition will improve.
Of the six patients with a postneonatal onset, all presented between 2 and 12 months of age with increased work of breathing and crackles on auscultation of the chest, after an apparently symptom-free period earlier in life. The reason these infants do not present at birth is difficult to explain. At birth, additional stress is placed on interstitial clearance mechanisms as the large volume of foetal lung liquid that occupies the alveolar compartment during gestation is absorbed from the lung lumen to make way for air-breathing 23. Whilst some alveolar fluid is cleared via the lung lymphatics, resulting in an increase in lung lymph flow at birth, most of lung liquid reabsorbed from the alveolar space is thought to pass directly into the foetal circulation 24. This lack of reliance of the pulmonary lymph channels at birth may explain why the majority of patients with PPL did not develop respiratory failure in the neonatal period. There is a paucity of information regarding lymphatic development in infancy that can assist in explaining why a number of infants present with symptoms for the first time at a few months of age, some of whom worsen during the first few months after presentation 15, 25. There are few chronic clinical entities that mimic these findings; the differential diagnosis includes: pulmonary aspiration syndromes, interstitial lung disease and indolent/recurrent pulmonary infection.
In addition to significant respiratory symptoms and recurrent lower respiratory infections, the infants in the current study had significant growth failure during the first year of life, which corrected fully by 3 yrs of age. There was no clinical evidence of malabsorption in these infants, and the failure to gain weight was ascribed to decreased intake and increased energy expenditure associated with respiratory effort and infections. The repeated lower respiratory infections in the studied patients were assumed to be secondary to mechanical factors (e.g. poor lung compliance) related to retention of interstitial fluid. The mild immune deficiencies detected in two patients may have contributed to the recurrent infections, but other patients did not have evidence of immune dysfunction. Since gastroesophageal reflux has been associated with other chronic respiratory diseases 26, two patients underwent Nissen fundoplication for documented reflux that was unresponsive to medical therapy. There was no improvement in respiratory condition after surgery.
A clinical diagnosis of PPL can be made in full-term neonates presenting with respiratory distress, pleural effusions (particularly if chylous) with or without generalised oedema. In older infants, the diagnosis may be harder to determine because symptoms are likely to be more subtle and may be similar to other causes of persistent tachypnoea in infancy 27. The diagnosis can be assisted by radiological findings and confirmed by lung biopsy. The radiological findings in the studied patients are similar to those described previously by others 2, 11, 15, 20, 25. The progression of generalised hazy infiltrates in the neonatal period to a more perihilar interstitial pattern with hyperinflation was seen consistently. A recent report suggests that magnetic resonance imaging may be useful for the imaging of pulmonary lymphangiectasia 18. The decision to submit patients to lung biopsy was determined by the uncertainty of the diagnosis. In the patient who did not have a lung biopsy, the diagnosis of PPL was made from a presentation of generalised oedema and chylothorax. In the patients who had repeat lung biopsy, the pathological findings changed over time. The first biopsies indicated nonspecific changes that were probably secondary to an acute viral infection. The absence of lymphagiectatic changes on the first biopsies raises the possibility that viral infection may have been a trigger for subsequent lymphatic obstruction. The ability of viruses to induce structural changes in the lung is well documented 28; however, the current authors are not aware of any reports of damage to pulmonary lymphatics following viral infection.
From the current authors' experience, the pathological diagnosis of PPL from a lung biopsy can be made with confidence, since minimal evidence of lymphangiectasia artefacts that could be ascribed to biopsy technique were seen. The biopsy samples from infants with clinical and radiological evidence of PPL showed severe lymphatic dilation in all three lymphatic compartments and were readily differentiated from the occasional isolated lymphangiectasia seen in the control samples from children's lungs.
The widely held view that this condition has a fatal outcome was moulded by earlier reports of primary pulmonary lymphangiectasia in the pre-ventilation era of neonatal intensive care. The current authors conclude that the recent descriptions of this condition as having a “dismal” 15, “usually lethal” 29 or “very poor” 30 prognosis do not take account of the contribution of coexisting disease to outcome in many of these patients or the recent advances in neonatal care that allow survival in neonates with primary pulmonary lymphangiectasia. The current authors' experience, as well asthat described in numerous case reports, suggests that survival can be anticipated in neonatal onset primary pulmonary lymphangiectasia without additional anomalies, and that the prognosis is excellent for the postneonatal onset form of primary pulmonary lymphangiectasia. The limited observation of a number of these studied patients over time suggests that, although not normal, lung function does not worsen with advancing age. However, the long-term prognosis and lung function in adult survivors with primary pulmonary lymphangiectasia has not been determined.
- Received February 4, 2004.
- Accepted May 20, 2004.
- © ERS Journals Ltd










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}