





Abstract
Acute respiratory distress syndrome (ARDS) is a disease of multifactorial etiology characterised by rapid development of severe diffuse and nonhomogenous inflammation of the pulmonary lobules causing life-threatening hypoxaemic respiratory failure. The current authors tested a therapeutic intervention on a previously defined pathophysiological model of ARDS. The model was defined by investigating, during the natural history of ARDS, the relationship among the three fundamental elements of a disease process pathogenesis, structural alterations, and functional consequences. In these studies, the present authors provided biological and morphological evidence indicating that ARDS patients failing to improve after 1 week of mechanical ventilation (unresolving ARDS) have intense and protracted (dysregulated) pulmonary and systemic inflammatory and neo-fibrogenetic activity.
Nuclear factor-κB and the glucocorticoid receptor have diametrically opposed functions in regulating inflammation. This chapter will review recent data indicating that poor outcome in acute respiratory distress syndrome might be related in part to failure of the activated glucocorticoid receptors to downregulate the transcription of inflammatory cytokines despite elevated levels of circulating cortisol. In a small randomised study of patients with unresolving acute respiratory distress syndrome, the current authors have shown that prolonged glucocorticoid supplementation improved all aspects of glucocorticoid receptors function and enhanced glucocorticoid-mediated anti-inflammatory action by interfering with nuclear factor-κB activation.
- acute respiratory distress syndrome
- biology
- glucocorticoid
- glucocorticoid receptor
- nuclear factor-κB
- outcome
This work was supported by the Baptist Memorial Health Care Foundation and the Assisi Foundation of Memphis.
Acute respiratory distress syndrome (ARDS) is a term applied to a clinical condition of multifactorial etiology associated with a relatively specific morphological lesion termed “diffuse alveolar damage” 1. This chapter will review studies that have tested a therapeutic intervention (glucocorticoid (GC) treatment) on a previously defined pathophysiological model of ARDS 2. The model was developed by investigating during the longitudinal course of ARDS, the relationship between the three fundamental elements of a disease process pathogenesis, structural alterations, and functional consequences 3.
At presentation, (early) ARDS manifests with severe, diffuse, and nonhomogenous acute host inflammatory response (HIR) of the pulmonary lobules leading to a breakdown in the barrier and gas exchange function of the lung. Injury to the alveolo-capillary membrane (ACM) causes flooding of the airspaces with protein-rich neutrophilic oedema fluid, resulting in severe gas exchange and lung compliance abnormalities 4. While a regulated inflammatory response is critical to survival 5, a major predictor of poor outcome in ARDS patients is persistence of pulmonary and systemic inflammation after 1 week of lung injury 6, 7. Failure to downregulate the production of inflammatory mediators (dysregulated HIR) is associated with maladaptive lung repair and inability to improve ACM permeability, gas exchange, and lung mechanics over time.
The lung injury score (LIS) quantifies the physiological respiratory impairment in ARDS through the use of a four-point score based on the levels of positive end-expiratory pressure, arterial oxygen tension: inspiratory oxygen fraction, the static lung compliance, and the degree of infiltration present on chest radiograph 8. Patients failing to improve the LIS or its components by day 7 of ARDS (nonimprovers) have a poor outcome 9–11. The present authors have previously reported that patients meeting predefined criteria for unresolving ARDS (LIS on day 7 of ARDS ≥2.5 and <1‐point reduction from day 1 of ARDS) have a mortality rate in excess of 80% (table 1⇓) 12.
Definitions of resolving and unresolving acute respiratory distress syndrome (ARDS)
Model of translational research
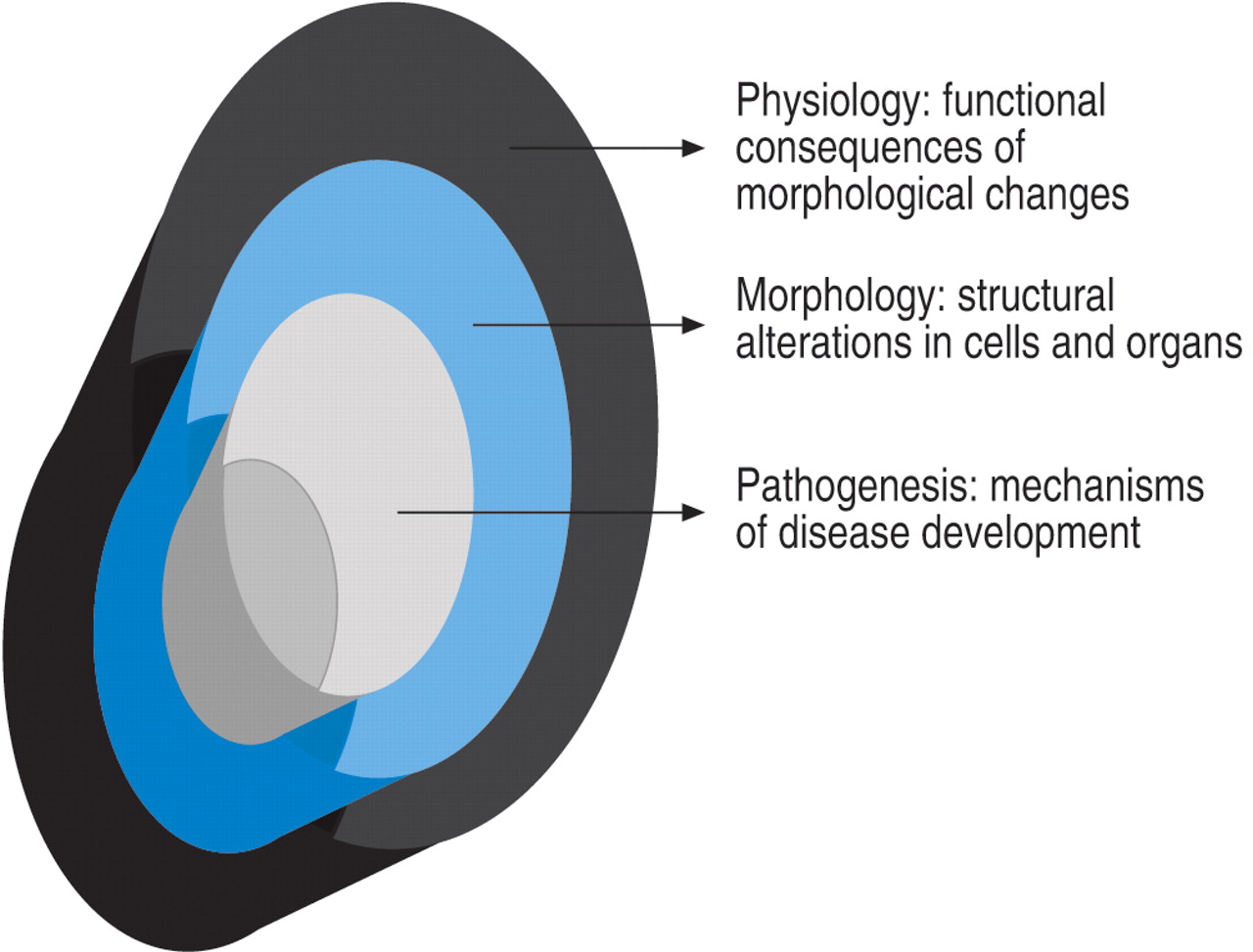
Because there is no animal model to study the progression of ARDS, translational clinical research has an important role to play for advancing understanding in this field. At the University of Tennessee the current authors have followed a “holistic” level of inquiry in constructing a pathophysiological model of ARDS attempting to fit pathogenesis (biology) with morphological (pathology) and clinical (physiology) findings observed during the longitudinal course of the disease (fig. 1⇓) 3. Most importantly, the current authors have attempted to define the differences over time between patients with an adaptive (resolving ARDS) versus maladaptive (unresolving ARDS) reparative response, and the time span of disease reversibility (prior to reaching end-stage disease) that is potentially amenable to anti-inflammatory treatment. Patients failing to improve after 1 week of ARDS onset (unresolving ARDS) were also studied to investigate the effect of prolonged anti-inflammatory (GC) treatment. In relation to the treatment investigation, it was assumed that the closer the treatment intervention was to the core pathogenetic mechanisms of the diseases, the more likely treatment would affect all “layers” of the disease process (fig. 1⇓). In this context, a positive or negative biological and physiological response to treatment was used to test the pathogenetic relevance (central versus peripheral) of the factor or pathway affected by treatment.

Disease elements.
Over the last decade scientific understanding of the intermediary events that occur between the reception of a biological signal at the cell membrane, and the eventual conversion of that signal to a change in gene expression at the nuclear level (i.e. signal transduction) has grown immensely 13. It is now recognised that two cellular signalling pathways are central to the regulation of inflammation, the stimulatory nuclear factor-κB (NF-κB) and the inhibitory glucocorticoid receptor (GR)‐α‐mediated signal transduction cascades. In unstimulated cells, both NF-κB and GR are predominantly sequestered in the cytoplasm.
Nuclear factor-κB
NF-κB is recognised as the central transcription factor that drives the inflammatory response to insults. NF-κB activation is an essential step in the experimental development of neutrophilic lung inflammation 14–16. NF-κB is found in essentially all cell types and is involved in activation of an exceptionally large number of target genes (over 100) 17. NF-κB is a heterogenous collection of dimers, composed of various combinations of the NF-κB/Rel family. The p65:p50 heterodimer was the first form of NF-κB to be identified and is the most abundant in most cell types 17. NF-κB is maintained in an inactive form by sequestration in the cytoplasm through interaction with inhibitory proteins (IκBs; most important being IκBα) (fig. 2⇓) 18. Activation of NF-κB is a rapid, immediate early event that occurs within minutes after exposure to a relevant inducer, including innate immunity stimulating molecules (e.g. lipopolysaccharide), double-stranded deoxyribonucleic acid (DNA), physical and chemical stresses, and inflammatory cytokines (e.g. tumour necrosis factor (TNF)‐α and interleukin (IL) ‐1β). In response to these various stimuli, the latent NF-κB/IκB complex is activated by phosphorylation and proteolytic degradation of IκB, with exposure of the NF-κB nuclear localisation sequence 17. Proteolytic degradation of IκB is an irreversible step in the signalling pathway that constitutes a commitment to transcriptional activation 17.

{kind=link}
{kind=link}
Interaction between nuclear factor (NF)-κB and the activated glucocorticoid receptor (GR). When cells are stimulated by inflammatory signals, specific kinases phosphorylate the inhibitory protein IκB and cause its rapid degradation. The activated form of NF-κB then moves to the nucleus initiating the transcription of the messenger ribonucleic acid (mRNA) of inflammatory cytokines, chemokines, cell adhesion molecules (e.g. intercellular adhesion molecule (ICAM)), and inflammation-associated enzymes (cyclooxygenase (COX), phospholipase A2 (PLA2), inducible nitric oxide synthase (iNOS)). Cortisol or exogenous glucocorticoids (GCs) freely cross into the cytoplasm and bind to their specific GC receptors (GRα) to form the activated receptor (GC-GRα). GC-GRα complexes may influence NF-κB activity in 5 major ways: 1) physically interacting with the p65 subunit with formation of an inactive (GC-GRα/NF-κB) complex; 2) inducing the synthesis of the inhibitory protein IκBα via interaction with glucocorticoid-responsive element deoxyribonucleic acid (DNA) in the promoter of the IκB gene; 3) blocking degradation of IκBα via enhanced synthesis of interleukin (IL)-10; 4) impairing tumour necrosis factor (TNF)‐α‐induced degradation of IκBα; and 5) competing for limited amounts of GR co-activators such as CREB-binding protein and steroid receptor coactivator‐1. GC-GRα may also decrease the stability of mRNA of several pro-inflammatory cytokines and other molecules. Products of the genes that are stimulated by NF-κB activate this transcription factor. Thus, TNF‐α and IL‐1β both activate and are activated by NF-κB, by forming a positive regulatory loop that amplifies and perpetuates inflammation.
The liberated NF-κB then translocates into the nucleus and binds to promoter regions of target genes to initiate the transcription of multiple cytokines including TNF‐α, and the interleukins IL‐1β, IL‐2, IL‐6, chemokines such as IL‐8, cell adhesion molecules (e.g. intercellular adhesion molecule‐1, E‐selectin), interferon, receptors involved in immune recognition such as members of the major histocompatibility complex, proteins involved in antigen presentation, receptors required for neutrophil adhesion and migration, and inflammation-associated enzymes (cyclooxygenase, phospholipase A2 (PLA2), inducible nitric oxide synthase) 18, 19. Products of the genes that are stimulated by NF-κB activate this transcription factor. Thus, TNF‐α and IL‐1β both activate and are activated by NF-κB, by forming a positive regulatory loop that amplifies and perpetuates inflammation 20. NF-κB also operates in conjunction with other transcription factors, including activator protein‐1 (AP‐1) 21.
Glucocorticoid receptor
Peripherally generated TNF‐α, IL‐1β, and IL‐6 activate the hypothalamic-pituitary-adrenal (HPA) axis independently at some or all of its levels 22, 23. The HPA axis responds in a graded manner to greater intensities of stress with increased production of adrenocorticotropic hormone (ACTH) and GCs. Due to their hormonal and lipophilic nature, GCs pass freely through the cell membrane. GCs exert most of their effects by activating ubiquitously distributed (2,000 to 30,000 per cell) cytoplasmic heat shock protein-complexed GR with formation of GC-GR complexes 24. It is now appreciated that the GC-GR complexes modulate transcription in a hormone-dependent manner by binding as a dimer to glucocorticoid-responsive elements (GREs) located in the promoter regions of GC responsive genes and by interfering with the activity of other transcription factors such as NF-κB on genes regulated by these factors 25. As a dimer and/or a monomer, GR-mediated transcriptional interference is achieved by five important mechanisms (fig. 2⇑): 1) physically interacting with the p65 subunit and formation of an inactive (GR-NF-κB) complex 24; 2) by inducing the transcription of the inhibitory protein IκBα gene 24, 26, 27; 3) by blocking degradation of IκBα via enhanced synthesis of IL-10 28–30; 4) by impairing TNF‐α‐induced degradation of IκBα 31, 32 and 5) by competing for limited amounts of GR co-activators such as CREB-binding protein and steroid receptor coactivator‐1 33. In addition to the transcriptional modulation described above, GCs also influence the processing of messenger ribonucleic acid (mRNA) and translation of proteins probably through transactivation or transrepression of genes that regulate mRNA stability and translation 34.
Inflammation-associated glucocorticoid inadequacy/resistance
GCs as end-effectors of the hypothalamic-pituitary-adrenal axis are the most important natural inhibitors of inflammation 35. However, endogenous GCs are not always effective in suppressing life-threatening systemic inflammation, even though the degree of cortisolemia frequently correlates with severity of illness and mortality rate 36–39. Unquestionably, the elevation of GC secretion in nonsurvivors is inadequate to meet the needs of the concurrent inflammatory response and its adverse systemic effects. Failure to suppress inflammation could be due to tissue resistance to GCs, inadequacy of the level and duration of endogenous GC elevation to suppress an HIR gone awry, or both 40.
The concept of acquired GC resistance was first introduced by Kass and Finland 41 in 1957. These investigators suggested that increased blood cortisol levels in patients with sepsis may reflect a block to steroidal activity or transport, as a consequence of the infection. In this situation, a small increase in blood levels with a low (equal or less than physiological) dose of exogenous GCs was believed to be sufficient for facilitating the passage of steroids into host cells 41. GR-mediated resistance was originally described as a primary inherited familial syndrome 42, 43 and was recently recognised as an “acquired” condition. Among others, acquired immune tissue-specific GR resistance has been described in patients with asthma 44–47, acquired immunodeficiency syndrome (AIDS) 48 and severe sepsis 49.
Recent in vitro studies have shown that cytokines may induce resistance to GCs by reducing GR binding affinity to cortisol and/or GREs 50–52. Such abnormalities of GR function were demonstrated in T‐cells incubated with a combination of IL‐2 and IL‐4 51. IL‐1, IL‐6, and interferon (IFN)‐γ 50 or IL-13 52. GC resistance was induced in a cytokine concentration-dependent fashion and was reversed by the removal of cytokines 51. GR-mediated resistance in the presence of systemic inflammation was also studied in experimental models of sepsis and sepsis-induced ARDS 49, 53, 54. In a sheep model of sepsis-induced ARDS, maximal binding capacity of GR decreased continuously after endotoxin infusion, while there was a marked elevation of cortisol levels 53. The reduced GR binding correlated negatively (r=−0.87, p<0.01) with PLA2 activity, a gene that is stimulated by NF-κB. In a rat model of septic shock, GR blockade by mifepristone (RU 486) exacerbated the physiological and pathological changes induced by endotoxemia 54. PLA2 activity in rats with 80% GR blockade was more marked than in those with 50% GR blockade 54. Monocytes of patients with sepsis developed near total GC resistance in vitro, when cytokines, especially IL‐2, were added 49.
Several inflammatory cytokines, including TNF‐α, IL‐1β, and IL‐6 activate NF-κB 55. It has been proposed that when cytokine-activated NF-κB forms protein-protein complexes with activated GR, the availability and activity of effective GR molecules are reduced 24, 47. This functional reduction in GR availability is associated with decreased GR-GRE DNA binding and GC-mediated anti-inflammatory activity 24, 47.
Longitudinal studies of biomarkers of host inflammatory response in acute respiratory distress syndrome
The current authors previously reported data to support a single “hit” model for ARDS progression, where degree and duration of the HIR determined the adaptive versus maladaptive evolution of the reparative process and final outcome. In a series of studies 56–59 the current authors have shown that patients with ARDS failing to improve in the first week of mechanical ventilation (unresolving ARDS) had biological and morphological evidence of intense and protracted pulmonary and systemic inflammatory and neo-fibrogenetic activity. Over time, patients with unresolving ARDS had persistent and exaggerated elevation in plasma and bronchoalveolar lavage (BAL) levels of TNF‐α, IL‐1β, IL‐6, IL‐8, 56–59 soluble intercellular adhesion molecule‐1 (sICAM‐1) 58 and procollagen aminoterminal propeptide type I (PINP) and type III (PIIINP) 60. During the first week of ARDS, pro-inflammatory cytokine levels declined in all survivors, while levels remained persistently elevated in all nonsurvivors. Recent data from the present authors' group indicate that cytokine levels reflected true biological activity 61. Furthermore, histological findings of open lung biopsies obtained in patients with unresolving ARDS (day 15±7 of mechanical ventilation) provided morphological evidence of persistent activation of the HIR. Histological findings in previously spared pulmonary lobules included new injury to the endothelial and epithelial surfaces with associated intravascular coagulation and extravascular fibrin deposition 6, 7. Histological findings in previously involved pulmonary lobules included progressive fibroproliferative obliteration with transformation of the initially fibrinous exudate into myxoid connective tissue matrix and eventually into dense acellular fibrous tissue 6, 7. Histological differences between survivors and nonsurvivors placed advanced pulmonary fibrosis with acellular fibrosis and loss of alveolar architecture at the upper boundary of disease reversibility 6.
Glucocorticoid treatment of unresolving acute respiratory distress syndrome
Large randomised studies have previously shown that a short course (≤24 h) of high-dose methylprednisolone (MP) in early ARDS is ineffective 10, 62–64. Table 2⇓ shows the differences between the older trials in early ARDS and the newer one in unresolving ARDS. The reference of Meduri 65 provides an historical review of GC treatment in sepsis and ARDS in relation to the evolving pathophysiological understanding of systemic inflammation. Because the half-life of MP is ∼180 min, a sustained pharmacological effect in life threatening protracted lung inflammation (i.e. status asthmaticus, Pneumocystis carinii pneumonia, etc.) can be achieved only with prolonged administration aimed at disease resolution. In a rat model of butylated hydroxytoluene-induced acute lung injury, GC administration was shown to be effective in decreasing lung collagen and oedema formation as long as treatment was prolonged; while withdrawal rapidly negated the positive effects of therapy 66–68. Two clinical studies demonstrated that premature discontinuation of prolonged GC administration in unresolving ARDS was associated with physiological deterioration that resolved with re-institution of treatment 69, 70. Moreover, studies suggest that premature discontinuation of GC therapy may not only lead to loss of the early treatment benefits, but may in fact be harmful. Indeed, in another study the cytokine response to lipopolysaccharide challenge in humans was significantly enhanced by a prior (12–144 h) short course of GCs 71. This observation may explain the differences in infection-related mortality between studies utilising a short course (24 h) 63 versus a prolonged course of methylprednisolone treatment 12, 72.
Comparison of old and new trials investigating methylprednisolone use in acute respiratory distress syndrome (ARDS)
Similar to the original reports from Ashbaugh and Maier 69 and Hooper and Kearl 70 the current authors investigated the use of prolonged MP administration in patients with unresolving ARDS. The current authors and others have reported significant improvement in lung function during prolonged MP administration in medical 6, 70, 73, 74 and surgical 69, 75 patients with unresolving ARDS and have found that survival correlated with improvement in lung function. The current authors' studies 6, 73 defined improvement in lung function as a reduction in LIS 8 of at least one point after 10 to 14 days of MP treatment. In phase II trials involving 34 patients, mortalities of 17% were reported in 29 patients who improved lung function (responders) and 100% in 5 nonresponders 6, 73. Additional findings of phase II trials are shown in ⇓tables 3 and 4⇓.
Phase II studies evaluating methylprednisolone in unresolving acute respiratory distress syndrome (ARDS). Findings at initiation of methylprednisolone treatment
Phase II studies evaluating methylprednisolone in unresolving acute respiratory distress syndrome (ARDS). Findings during methylprednisolone treatment
Most recently the present authors completed a prospective, randomised, double-blind, placebo-controlled trial designed to evaluate the efficacy of prolonged MP treatment (MPT) in patients with unresolving ARDS 72. Methylprednisolone or placebo was given daily as intravenous push every 6 h (one-fourth of the daily dose) and changed to a single per os dose when oral intake was restored. The MP dosage regimen is shown in table 5⇓. If the patient was extubated prior to day 14, treatment was advanced to day 15 of drug therapy and tapered according to schedule 72. The protocol contained: 1) a provision for blindly crossing over patients who did not improve LIS by at least 1 point after 10 days of treatment or earlier crossover in patients with life-threatening deterioration in gas exchange and; 2) procedures for infection surveillance, including weekly bronchoscopy with bilateral BAL 72. Because MP blunts the febrile response to an infection, this latter intervention was essential for minimising the random variation generated by the potential morbidity and mortality of untreated nosocomial infections. The study was designed as a sequential phase III clinical trial and projected to recruit 100 patients. The decision to end the trial was made when the test statistic exceeded the upper boundary of the triangular test of Whitehead, and the null hypothesis was rejected at a significance <0.05 and a power >0.95.
Methylprednisolone treatment in unresolving acute respiratory distress syndrome
The two groups were similar at study entry. After ten days, all patients in the MPT group improved (≥1 point reduction in LIS) versus 2 of 8 (25%) in the placebo group. Intensive care unit and hospital-associated mortality were significantly reduced: 0% versus 62% (p=0.002) and 12% versus 62% (p=0.03), respectively. The small number of patients may have biased the estimate of the treatment effect. The rate of complications between the two groups was similar. During MP treatment, pneumonia frequently developed in patients without fever (44%). Therefore, infection surveillance, including weekly bilateral bronchoscopic BAL, was useful for early detection of pneumonia and other serious infections. None of the recognised and appropriately treated infections developing during MP therapy affected resolution of ARDS or clinical outcome. Since the study was completed, the present authors routinely use prolonged MPT in patients with unresolving ARDS with a physiological improvement observed in 60–80% of patients.
The therapeutic anti-inflammatory and anti-fibrotic efficacy of prolonged MPT was assessed with serial measurements of HIR biomarkers. MPT was associated with a rapid and sustained reduction in mean plasma and BAL TNF‐α, IL‐1β, IL‐6, IL‐8, sICAM‐1, IL‐1 receptor antagonist (IL-1ra), soluble TNF receptor 1 and 2 (sTNFR1 and sTNFR2), PINP and PIIINP, and with increases in IL-10 and in anti-inflammatory-to-pro-inflammatory cytokine ratios (IL-1ra/IL‐1β, IL-10/TNF‐α, IL-10/IL‐1β) 59, 60, 78. Placebo administration was not associated with reductions in HIR biomarkers. The current authors believe that failure of older trials investigating massive doses of MP in early ARDS (table 2⇑) was attributable to the short duration of administration and not to timing of administration.
Evidence of inflammation-associated glucocorticoid inadequacy/resistance in unresolving acute respiratory distress syndrome
Using an ex vivo model of systemic inflammation in ARDS the current authors investigated intracellular upstream and downstream events associated with DNA-binding of NF‐κB and GRα in the peripheral blood leukocytes (PBLs) (naive cells) obtained from a healthy volunteer (laboratory work conducted by F. Stentz, Dept of Medicine, Pulmonary and Critical Care Division, The University of Tennessee Health Science Center, Memphis, Tennessee, USA) 79. PBLs were incubated for 3 h with 98 plasma samples obtained longitudinally from 17 patients with ARDS before and after randomisation to either placebo (n=6) or MP (n=11). The cells were processed for fractionation into cytosolic and nuclear components, ribonucleic acid (RNA) extraction, and intracellular labelling. The primary objectives of these studies were to quantify the relationships among circulating levels of inflammatory cytokines TNF‐α and IL‐1β and HPA-axis hormones (ACTH and cortisol), and intracellular activities mediated by NF-κB (NF‐κB κb‐binding and transcription of TNF‐α and IL‐1β) and GRα (GRα binding to NF‐κB, GRα binding to GRE DNA, stimulation of inhibitory protein IκBα, and stimulation of IL-10 transcription).
In the observation period prior to randomisation, the biological and physiological characteristics of the MP and placebo groups were similar. Patients had persistent elevations in plasma levels of inflammatory (TNF‐α, IL‐1β, and IL‐6) cytokines and HPA-axis (ACTH and cortisol) hormones and similar severity of organ dysfunction scores. In PBLs exposed to patients' plasma GRα-mediated activities were essentially unchanged over time, while NF-κB κb-binding and transcription of TNF‐α and IL‐1β increased over time. The current authors' hypothesised that inadequate secretion of cortisol and/or immune tissue resistance to endogenous GCs might explain the observed failure of activated GR to suppress inflammation (progressive increase in NF‐κB‐mediated activities) in the presence of persistently elevated ACTH and cortisol levels.
Patients treated with MP had rapid, progressive and sustained reductions in plasma TNF‐α, IL‐1β, IL‐6, ACTH and cortisol levels over time. These were associated with parallel improvements in pulmonary and extrapulmonary organ dysfunction scores (previously reported in reference 72). Normal PBL exposed to plasma samples collected during MP versus placebo treatment also exhibited rapid, progressive significant increases in GRα-mediated activities (GRα binding to NF-κB, GRα binding to GRE DNA, stimulation of inhibitory protein IκBα, and stimulation of IL-10 transcription), and significant reductions in NF-κB κb-binding and transcription of TNF‐α and IL‐1β. With MPT, the intracellular relations between the NF‐κB and GRα signalling pathways changed from an initial NF-κB-driven and GR-resistant state to a GR-driven and GR-sensitive one.
The current authors interpret the responses observed during methylprednisolone treatment to support the concept of inflammation-dependent acquired glucocorticoid resistance in patients with unresolving acute respiratory distress syndrome. The findings also underscore the central role played by activated glucocorticoid receptor‐α in regulating inflammation and provide strong mechanistic evidence for the efficacy of prolonged methylprednisolone treatment in unresolving acute respiratory distress syndrome. A controlled trial is in progress to evaluate the effect of prolonged methylprednisolone treatment initiated within 72 h of acute respiratory distress syndrome development.
- acute respiratory distress syndrome
- biology
- glucocorticoid
- glucocorticoid receptor
- nuclear factor-κB
- outcome
- © ERS Journals Ltd
References










Jump To
- Article
- Abstract
- Model of translational research
- Nuclear factor-κB
- Glucocorticoid receptor
- Inflammation-associated glucocorticoid inadequacy/resistance
- Longitudinal studies of biomarkers of host inflammatory response in acute respiratory distress syndrome
- Glucocorticoid treatment of unresolving acute respiratory distress syndrome
- Evidence of inflammation-associated glucocorticoid inadequacy/resistance in unresolving acute respiratory distress syndrome
- References
- Figures & Data
- Info & Metrics