





Abstract
It was hypothesized that celltocell interaction between human alveolar macrophages (AM) and alveolar epithelium, might be an important factor leading to nitric oxide synthase‐2 (NOS2) messenger ribonucleic acid (mRNA) and protein expression by constituent cells of the alveolar wall and/or AM.
NOS2 mRNA and the protein expression patterns of human AM and alveolar epithelial cells type II (AECII) isolated from normal parts of lung resections of patients with pulmonary malignancies were determined. In addition, NOS2 mRNA expression in human AM cocultured with autologous AECII in the presence of proinflammatory cytokines interleukin (IL)‐1β, tumour necrosis factor (TNF)‐α, interferon (IFN)‐γ or lipopolysaccharide (LPS) was investigated. The effect of human surfactant protein‐A (SP‐A) on IFN‐γ‐mediated NOS2 mRNA expression in human AM was also studied.
Neither NOS2 mRNA nor protein could be detected in freshly isolated, unstimulated or cytokinestimulated AECII. In contrast, freshly isolated AM from bronchoalveolar lavage or lung tissue samples expressed immunoreactivity for NOS2 protein, but no NOS2 mRNA could be detected by reverse transcriptase polymerase chain reaction. All stimuli tested failed to induce NOS2 mRNA expression in human AM in vitro. Only AMAECII coculture in the presence of IFN‐γ led to NOS2 mRNA and protein expression. In situ hybridization of NOS2 mRNA on lung tissue explants and immunohistochemical staining of cytospin preparations of AMAECII cocultures demonstrated that NOS2 is expressed in AM but not in AECII. This coculture effect could not be reproduced by substitution of AECII with SP‐A.
These data give evidence of a regulatory network controlling human nitric oxide synthase‐2 expression in the lower respiratory tract.
- alveolar epithelial cells type II
- alveolar macrophages
- cytokines
- nitric oxide synthase‐2
- reverse transcriptase polymerase chain reaction
- surfactant protein A
This study was supported in part by a grant from the Deutsche Forschungs gemeinschaft (No. Mu 6623/5‐5). D.V. Pechkovsky is a recipient of a research fellowship from the European Respiratory Society and the Borstel Foundation.
Nitric oxide (NO) is a gas that can be detected in exhaled air and its increase is observed in several pulmonary disorders including tuberculosis, sarcoidosis, idiopathic pulmonary fibrosis (IPF), primary lung cancer, and adult respiratory distress syndrome (ARDS) 1–5. Exhaled NO has been shown to be an indirect marker of airway inflammation; alveolar macrophages (AM) and airway epithelial cells are assumed to be the main source of the elevated levels of NO 1–5.
Prolonged and enhanced NO production is thought to occur predominantly through the action of nitric oxide synthase type‐2 (NOS2). Although several investigators have shown that human AM can be induced to express NOS2 in vivo, their ability to express NOS2 and to generate NO in response to an in vitro stimulation is still under debate 6, 7. One reason for this controversy may arise from complex inflammatory processes within the alveoli responsible for NOS2 messenger ribonucleic acid (mRNA) expression, enzyme production, and NO generation by human AM. Despite numerous potential mechanisms, which can modulate NOS2 expression in the injured alveolar compartment, one common pathway is the formation of cytokine profiles within alveoli by constituent and recruited cells, including AM and T‐lymphocytes. More specifically, the accumulation of NOS2 mRNA and/or protein has been documented in AM or alveolar epithelium of patients suffering from pulmonary tuberculosis, sarcoidosis or IPF. This NOS2 expression is probably induced by proinflammatory cytokines such as tumour necrosis factor (TNF)‐α, interleukin (IL)‐1β, and interferon (IFN)‐γ 2, 8, 9. Either by direct production of NO or by expression of excessive amounts of bioactive substances, which in turn mediate NO production by both AM and alveolar epithelium, these cells may play an important role in lung injury and inflammation. Nevertheless, the cooperative contribution of alveolar epithelial cells type II (AECII) and AM to these processes is unclear.
The capabilities of human AM to control the NOS2 expression and NO production of the murine lung epithelial cell line LA‐4 by IL‐1β and TNF‐α has been demonstrated by Robbins et al. 10. It has also been shown that surfactant protein A (SP‐A), which is abundantly produced by AECII, induces NOS2 expression and NO production by rat AM 11. In contrast, more recently, the authors reported that human SP‐A itself did not affect production of NO or NOS2 protein expression in rat AM, but it significantly enhanced NOS2 protein production in IFN‐γ or lipopolysaccharide (LPS)/IFN‐γ stimulated cells 12.
The authors hypothesized that celltocell interaction between human AM and alveolar epithelium during inflammation might play a pivotal role in their NOS2 mRNA accumulation and protein expression. To test this hypothesis, human AM and AECII isolated from normal parts of lung resections of patients with pulmonary malignancies were examined, for their capabilities to express NOS2 mRNA and protein in primary culture. In addition the NOS2 expression patterns in human AM cocultured with autologous AECII, both in the absence or presence of proinflammatory cytokines (IL‐1β, TNF‐α, IFN‐γ) or LPS were determined. The effect of human SP‐A on IFN‐γ‐mediated NOS2 mRNA expression in human AM was also studied.
It was found that NOS2 mRNA and protein are absent in freshly isolated, unstimulated or cytokinestimulated AECII. In contrast, freshly isolated human AM from either bronchoalveolar lavage (BAL) or lung tissue samples expressed immunoreactivity for NOS2 protein, but no NOS2 mRNA could be detected by reverse transcription polymerase chain reaction (RTPCR). All stimuli tested failed to induce NOS2 mRNA expression in human AM in vitro. Only AMAECII coculture in the presence of IFN‐γ led to NOS2 mRNA accumulation and protein production in AM. This coculture effect could not be reproduced by substitution of the AECII with SP‐A.
These data give evidence of a regulatory network controlling human NOS2 expression in the lower respiratory tract.
Materials and methods
Specimens
BAL fluid samples were obtained from subjects who underwent bronchoscopy for diagnostic reasons. Five patients with normal chest radiographs and pulmonary function tests entered the study. BAL was performed as previously described 13. In addition, AM and AECII were isolated from 10 lung tissue samples, which were obtained from resected lung material. All subjects were smokers and had no respiratory tract infection within the last month. None were taking antibiotics or immunosuppressants at the time of investigation. Informed consent was obtained from all subjects. The study was approved by the appropriate review or medical ethics committees of the institutions involved.
Primary human alveolar epithelial cells type II
Samples were cut from the surgical specimens and used for a cell isolation procedure as described previously 14. In brief, lung tissue was sliced and slices were washed three times at 4°C in phosphate buffered saline (PBS; Gibco BRL, Paisley, Scotland) followed by an incubation in sterile dispase solution at 37°C for 45 min. After dispase digestion the lung tissue slices were cut into small, pipetable pieces, and thoroughly pipetted for 10 min. Crude tissue and cell suspensions were filtered through nylon gauze with meshes of 100, 50, and 20 µm. The resulting singlecell suspension was placed on Ficoll separating solution and centrifuged at 800×g for 20 min. The AECIIenriched cells from the interphase were incubated in 100 mm plastic dishes (NUNC, Wiesbaden, Germany) at 37°C in humidified air containing 5% carbon dioxide (CO2) for 15, 20 and 30 min with seeding of nonadherent cells on fresh dishes for each time interval to remove adherent cells (mostly AM and monocytes). To remove the remaining monocytes/macrophages and lymphocytes, antibodies against CD3 (OKT3, ECACC 86022706) and CD14 (ATCC HB246) were added and the antibodybinding cells were removed by antimouse immunoglobulin (Ig)‐G coated magnetic beads and Magnetic Activated Cell Sorting (MACS) system (Miltenyi Biotec, Bergisch Gladbach, Germany) as suggested by the supplier. Identity of AECII was confirmed by a modified Papanicolaou staining, their alkaline phosphatase activity, and SP‐A mRNA expression in RTPCR (see later). Cell purity was assessed by immunoperoxidase staining with monoclonal antibodies directed against CD3 and CD14 (Immunotech, Marseille, France) as previously described 15. Viability of the AECII after isolation was >97% as determined by trypan blue exclusion.
Alveolar macrophages
Human AM were isolated from BAL fluid samples and from the adherent cell fraction during AECII purification procedures. BAL fluid was filtered through a sterile nylon gauze filter, centrifuged at 500×g for 10 min at 4°C, and the cell pellet was washed with cold PBS. Cell differentials were determined by counting a minimum of 200 cells in a cytocentrifuge preparation (Cytospin II, Shandon, Pittsburgh, PA, USA) stained by HemacolorTM (E. Merck, Darmstadt, Germany). These cell suspensions contained >97% AM, 1–2% lymphocytes, and <1% neutrophil granulocytes. The viability of the cells was >95%. For isolation of AM from the same lungtissue sample as AECII, the adherent cells were harvested from plastic dishes with a cell scraper and vigorous pipetting after the addition of cold PBS. The cells were treated with mouse antibodies against human CD14 (ATCC HB246) and CD14positive (CD14+) cells were separated by MACS. After MACS purification, CD14+ cells were washed twice with PBS at 4°C. The cell suspension contained >85% AM, as assessed by specific morphology of the human AM, and by immunoperoxidase staining with monoclonal antibodies directed against CD14 as previously described 15. Contaminating cells were lymphocytes (5–10% CD3+ cells) and AECII (5–10% cells). Viability of the AM after isolation was >95%.
A549 cell line
A549 cells were a generous gift from T. Papadopoulos (University of Erlangen, Germany). This cell line was used as the positive control for NOS2 mRNA expression and protein production upon stimulation with proinflammatory cytokines and LPS. Experiments were performed with cells after seven passages, after thawing and inoculation in culture. Cells were grown on a 75cm2 tissue culture flask (NUNC) in culture medium (Roswell Park Memorial Institue (RPMI) 1640 medium (Gibco BRL) with 2 mM L‐glutamine, 10% heat inactivated foetal calf serum (FCS; Gibco BRL), 1% penicillin/streptomycin solution (Biochrom, Berlin, Germany), 1% sodium pyruvate solution (Biochrom) and 20 mM HEPES (Gibco BRL)) in a humidified atmosphere containing 5% CO2 at 37°C for 5 days. After this culture period, cells were removed from plastic surfaces by treatment with trypsin/ethylenediamine tetraacetic acid (EDTA) solution (BoehringerMannheim, Mannheim, Germany) (0.05/0.02% in PBS) for 10 min at 37°C, washed twice in PBS and resuspended in culture medium.
Cell cultures
To assess the effect of stimulation on NOS2 gene expression, AECII and AM (1×106 cells·mL−1) were treated with Salmonella minnesota LPS (1 µg·mL−1; K. Brandenburg, Research Centre Borstel, Borstel, Germany), recombinant TNF‐α (1–10 ng·mL−1; E. Schlick, Knoll AG, Ludwigshafen, Germany), recombinant human IFN‐γ (10–100 U·mL−1; Biotrend, Köln, Germany) or recombinant human IL‐1β (10–100 U·mL−1; Biotrend), and with a combination of these stimuli in collagen‐R coated (Serva, Heidelberg, Germany) 24 well plates (NUNC) at 37°C for 24 h. For timecourse experiments, cells were incubated in the presence or absence of LPS (1 µg·mL−1), TNF‐α (5 ng·mL−1), IFN‐γ (50 U·mL−1), or IL‐1β (50 U·mL−1) for 4, 8, 16, or 24 h. In separate experiments, AECII were cocultured with 2.5, 5, 10 or 15% of autologous AM either with or without LPS, TNF‐α, IL‐1β or IFN‐γ at the concentrations stated earlier. AM from BAL were stimulated with LPS (1 and 10 µg·mL−1), IFN‐γ (10–100 U·mL−1), or the combination of INF‐γ with SP‐A (1–100 µg·mL−1; J.R. Wright, Duke University, Medical Centre, Durham, NC, USA). Pulmonary SP‐A was isolated from lung lavage fluid from patients with alveolar proteinosis as previously described 16 and used in preparation containing <0.2 pg endotoxin·μg SP‐A−1. A549 were incubated with or without LPS (1 µg·mL−1), or cytokines for 24 h using the same protocol as for primary AECII or AM. After culture, cell viability always exceeded 95% in both AECII and AM as determined by trypan blue exclusion. Cellculture supernatants were removed and cells were lysed with Trizol reagent (Gibco BRL). Cell lysates were frozen and kept at −70°C before ribonucleic acid (RNA) isolation. For samples of RNA from freshly isolated cells, AM from BAL fluids, AM from lung tissue samples and AECII were subjected to RNA isolation procedures before culture, henceforth referred to as noncultured controls.
Reverse transcriptasepolymerase chain reaction
Total RNA was extracted from cells using Trizol according to the manufacturer's protocol. Equal amounts of total RNA from each sample were primed with oligo (dT)12–18 primer (Gibco BRL) and reversetranscribed with SuperScriptTM RNase H− reverse transcriptase (Gibco BRL) for 1 h at 37°C to produce complementary deoxyribonucleic acid (cDNA). PCR amplification of the cDNA (2.5 µL) was carried out using primer pairs specific for β‐actin and NOS2. To demonstrate that RNA samples from AECII were not significantly contaminated by RNAs from other type of cells (AM or lymphocytes) CD3‐ and CD14specific primers were also used. Sequences of the primers for analysis of mRNA's: NOS2 (forward): TCCGAGGCAAACAGCACA TTC; NOS2 (reverse): GGGTTGGGGGTGTGGTGATGT; β‐actin (forward): AGC GGG AAA TCG TGC GTG; β‐actin (reverse): CAGGGTACATGGTGGTGCC; CD3 (forward): GGCTGTCCTCATCCTGGCTATCAT; CD3 (reverse): ACTGGTTTCCTTGAAGGTGGCTGT; CD14 (forward): ACTCCCTCAATCTGTCGTTCGCTG; CD14 (reverse): CTGAAGCCAAGGCAGTTTGAGTCC; SP‐A (forward): TCTTTGGATGCCAAC TCA GC; SP‐A (reverse) CTTTATTCAGCTCAGGGGTG. All primers were synthesized by MWGBiotech (MWGBiotech AG, Ebersberg, Germany). Target cDNA was amplified using a threetemperature PCR and an automated thermocycler (Biometra, Göttingen, Germany) according to Reiling et al. 17 with primer pairs for NOS2, and according to Murray et al. 18 with primer pairs for CD3 and CD14, respectively. PCR conditions for β‐actin amplification included: 95°C for 1 min, 57°C for 1 min and 72°C for 1 min 30 s; and for SP‐A: 94°C for 1 min, 54°C for 1 min, 72°C for 1 min 30 s, and 72°C (terminal extension) for 15 min. The numbers of cycles were the same for NOS2, CD3, CD14, SP‐A, and β‐actin (35 cycles). PCR products (length: 462 bp, 517 bp, 341 bp, 622 bp, and 309 bp for NOS2, CD3, CD14, SP‐A, and β‐actin respectively) were electrophoresed on 1.5% agarose (Gibco BRL) gels and stained with ethidium bromide. Gel analysis was performed densitometrically with the “Gel Doc 2000” gel documentation system and “Quantity One 4.0.3” software (BioRad Laboratories, Hercules, CA, USA). To assure the identity of the PCRamplified fragments, the size of each amplified mRNA fragment was compared with deoxyribonucleic (DNA) standards (100 bp DNA Ladder; Gibco BRL) electrophoresed on the same gel. Additionally, the NOS2 specificity of the PCR product was verified by comparing their sequence data with published sequences. The NOS2specific primers used in the RTPCR were intronspanning and therefore genomic DNA contamination did not influence the PCR results 17.
Immunocytochemistry
AM and AECII (2.5×105 cells) were plated in duplicate chambers of an eightchamber glass slide (FlexipermSlide, W.C. Heraeus GmbH, Hanau, Germany) and cultured in culture medium, in the presence or absence of IFN‐γ (50 U·mL−1), or a combination of cytokines (TNF‐α (5 ng·mL−1), IFN‐γ (50 U·mL−1) and IL‐1β (50 U·mL−1)) for 18 h at 37°C. BAL cells were incubated under the same conditions. The cytocentrifuge slides were prepared with AECII, AM and BAL cell preparations without previous culture of the cells. Slides were washed in PBS and cells were fixed with 3.7% paraformaldehyde in PBS for 10 min. After fixation, slides were washed three times in PBS/0.1% saponin. Endogenous peroxidase was blocked by incubation with 0.3% hydrogen peroxide/0.02% sodium azide for 15 min, followed by a further incubation with 10% normal goat serum in PBS/0.1% saponin for 10 min. Between the various steps the slides were washed in PBS/0.1% saponin at room temperature. The slides were incubated with a murine monoclonal antihuman NOS2 antibody (Transduction Laboratories, Lexington, KY, USA) at 4 µg·mL−1 in PBS/0.1% saponin for 2 h at room temperature. Control slides were incubated in PBS/0.1% saponin alone. After washing and incubation with a biotinylated goat antimouse IgG antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) for 30 min, and with streptavidin horseradish peroxidase (Zymed Labs, South San Francisco, CA, USA) for 30 min, the slides were treated with diaminobenzidine in PBS, counterstained with haematoxylin, mounted in glycerol gelatine and coverslipped. A549 cells stimulated with IFN‐γ (100 U·mL−1) and a mixture of cytokines for 18 h were used as positive controls. Slides with the primary antibodies omitted were used as negative controls.
Western blot analysis for nitric oxide synthase‐2
AECII or A549 cells (5×106) that had been incubated with or without appropriate stimuli were removed from flasks by treatment with trypsin/EDTA solution for 10 min at 37°C, washed in PBS and resuspended in 0.2 mL of a lysis buffer (50 mM Tris HCl, pH 7.6/100 mM NaCl/2 mM EDTA/2 mM EGTA/0.1% of Triton X/1% of proteaseinhibitor cocktail (Sigma, St. Louis, MO, USA)) and lysed for 30 min on ice. Cell debris was removed by centrifugation at 13000×g for 15 min. NOS2 protein was enriched by adenosine 2′5′diphosphateagarose (Sigma, Deisenhofen, Germany). Aliquots of this enriched fraction were resolved by sodium dodecylsulphate gel electrophoresis (SDSPAGE) (10% acrylamide) and transfered onto nitrocellulose membranes (Amersham Pharmacia biotech, Little Chalfont, UK) by using a semidry technique (Hoefer SemiDry Transfer Unit, Amersham Pharmacia biotech). The membranes were labelled with monoclonal mouse antiNOS2 antibodies, as suggested by the supplier (Transduction Laboratories, Lexington, KY, USA). The blots were developed using the enhanced chemiluminescence (ECL, Amersham Pharmacia) technique according to the manufacturer's instructions.
In situ hybridization
Paraffinembedded lungtissue samples were cut from the same surgical specimens as described earlier and used for in situ hybridization (ISH). These tissue samples showed normal architecture with few intraalveolar macrophages and oedema. The cDNA probe corresponding to the NOS2 gene was produced by PCR as described earlier, filtered through CentriSep spin columns (Applied Biosystems, Foster City, CA, USA), and labelled with digoxigenin (Dig) following the manufacturer's instructions (DigHighPrime, Roche, Germany). After deparaffinization, ISH was carried out overnight and, after washing at high stringency, detection was performed by application of AntiDig/alkalinephosphataseconjugate and newfuchsin as a substrate for the alkaline phosphatase 19. For the negative control, sections were hybridized with the hybridization buffer in the absence of labelled cDNA probes. The specificity of the hybridization signal was confirmed by treating preparations with ribonuclease (RNase) before hybridization.
Nitrite/nitrate determination
Total NOproducts were determined by a Nitrate/Nitrite colourimetric assay kit (Cayman Chemical, Ann Arbor, MI, USA) according to the suggestions of the supplier. This test employs a Griess reaction after enzymatic conversion of NO3− into NO2−.
Statistical analysis
Data are expressed as mean±sem. Statistical comparisons were made by analysis of variance (ANOVA) with post hoc Fisher's protected least significant difference (PLSD) for each agent separately. Probability values were considered significant if they were <0.05.
Results
Characteristics of isolated alveolar epthelial cells and alveolar macrophages
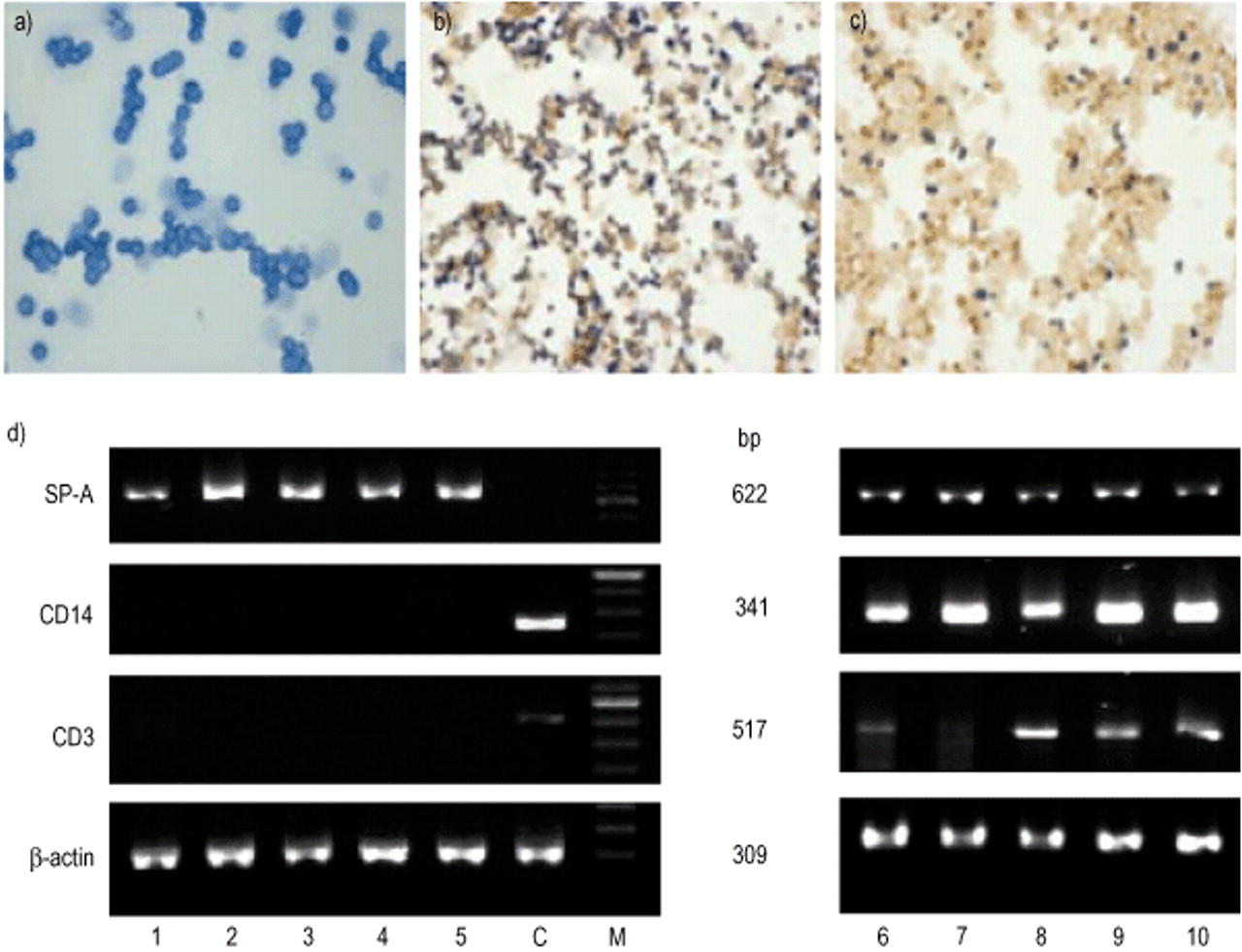
To minimize cellular contamination, human AECII isolated by three steps of purification, as described in the Materials and methods section, were used. After the final step of MACS purification, the AECII preparations included in this report were free of CD14+ and CD3+ cells as determined by immunocytochemistry. A total of 97±1.9% of cells were identified as AECII by the presence of dark blue inclusions as revealed by modified Papanicolaou staining and 94±1.1% of cells were positive for alkaline phosphatase (fig. 1a and 1b⇓). All RNA samples isolated from these AECII preparations contained SP‐A mRNA but no CD3 and CD14 mRNA as determined by RTPCR (fig. 1d⇓).

Photomicrographs of freshly isolated human alveolar epithelial cells type II (AECII) stained with a) modified Papanicolaou stain, and b) for alkaline phosphatase (AP), and counterstained with 0.1% neutral red. c) Alveolar macrophages (AM) freshly isolated from lung tissue samples and stained for expression of AP. d) The results of reversetranscriptase polymerase chain reaction (RTPCR) product analysis with ribonucleic acid (RNA) from five freshly isolated AECII and AM preparations (lanes 1–5 and 6–10 respectively) and specific primers for surfactant protein A (SP‐A), CD14, CD3, and β‐actin. Lane C shows the results of RTPCR with RNA isolated from one noncultured human bronchoalveolar lavage cell preparation used as positive control for CD3 and CD14, and as negative control for SP‐A.
The autologous AM, which were obtained from the same lung tissue samples and purified by MACS “positive” selection, contained 85% of CD14+cells, and were contaminated with 6.4±4.3% of CD3+cells and 7.8±3.6% of AECII. These data were confirmed by immunoperoxidase staining with monoclonal antibodies directed against human CD14 and CD3 surface antigens (data not shown), alkaline phosphatase staining (fig. 1c⇑) and by RTPCR for CD3 and SPA mRNA (fig. 1d⇑). Additional evaluation of these cell preparations under a light microscope revealed that CD3+ cells and AECII displayed a strong capability to adhere on the AM surface (data not shown), and therefore contaminated the CD14+ cell populations.
Cells retrieved from BAL samples disclosed higher percentages of AM (93.3±2.7%, n=5) which were contaminated with 3.2±1.8% lymphocytes, 1.8±0.9% neutrophils, but free of AECII as shown by SP‐A mRNA analysis (fig. 1d⇑), henceforth referred to as BALAM. These cell populations were not purified, so that introduction of artefacts in experimental conditions was avoided.
Nitric oxide synthase type‐2 messenger ribonucleic acid expression in aveolar epithelial cells type II and alveolar macrophages isolated from lung tissue samples
RTPCR with RNA samples from freshly isolated noncultured AECII as well as from cultured unstimulated and cytokinestimulated cells did not reveal the presence of any NOS2 mRNA (fig. 2a⇓). NOS2 gene expression could not be detected in cells stimulated with IFN‐γ (10, 50, and 100 U·mL−1), with LPS (1 µg·mL−1), TNF‐α (1, 5, and 10 ng·mL−1) or IL‐1β (10, 50, and 100 U·mL−1) for 4, 8, 16, and 24 h, and with the combination of the three cytokines at the same time periods (data not shown). All RNA samples isolated from these cultures were positive for SP‐A and intensities of the bands were not different in unstimulated and cytokinestimulated cells over all time periods of culture (data not shown). In AM isolated from lung tissue samples there was no detectable NOS2 mRNA in noncultured, unstimulated or in cultured cytokinestimulated cells at different time periods and stimulation protocols (fig. 2b⇓).

Nitric oxide synthase‐2 (NOS2) messenger ribonucleic acid (mRNA) analysis by reversetranscriptase polymerase chain reaction (RTPCR) of noncultured (lane 0) and cultured alveolar epithelial cells type‐II (AECII), alveolar macrophages (AM), and A549 cells. Cells were cultured without (lane 1) or with tumour necrosis factor (TNF)‐α (5 ng·mL−1; lane 2), interferon (IFN)‐γ (50 U·mL−1; lane 3) or interleukin (IL)‐1β (50 U·mL−1; lane 4) for 24 h. a) Representative images of NOS2 mRNA expression in AECII are from one of five identical experiments, and b) from one of ten identical experiments with AM isolated from lung tissue samples. c) Time/course experiment after the seventh passage shows the effect of lipopolysaccharide (LPS) or cytokinestimulation on NOS2 mRNA expression by A549 cells after 4, 8, and 24 h. C: positive control.
In contrast to AECII and AM, the stimulation of A549 cells with IFN‐γ or TNF‐α for 24 h resulted in an upregulation of NOS2 mRNA expression (fig. 2c⇑). Stimulation of AM, AECII, or A549 cells with LPS alone (1 or 10 µg·mL−1) did not lead to the upregulation of NOS2 mRNA expression in any of the experimental conditions tested (fig. 2c⇑).
Another pattern of NOS2 mRNA expression was found when AECII were stimulated with LPS or cytokines for 4, 8, 16 and 24 h in the presence 10% of autologous AM. As shown in figure 3⇓, stimulation with IFN‐γ (50 U·mL−1) and IL‐1β (50 U·mL−1) leads to a statistically significant upregulation of NOS2 mRNA expression in a dose and timedependant manner. There were lower NOS2 mRNA expression levels after stimulation with 10 U·mL−1 compared to 50 U·mL−1 or 100 U·mL−1 of IFN‐γ at the earlier time points of AMAECII cocultures (data not shown). Stimulation with 100 U·mL−1 resulted in lower message levels compared to 50 U·mL−1 of IFN‐γ after 24 h, and the increase of cytokine concentrations >100 U·mL−1 disclosed an inhibitory effect on NOS2 expression (data not shown). Therefore a concentration of 50 U·mL−1 IFN‐γ and a 24‐h cellculture period for further experiments with AMAECII cocultures was used. No significant amounts of NOS2 mRNA were found in freshly isolated cells, in cells without additives and in cells stimulated with TNF‐α or LPS (fig. 3⇓). Thus, the level of NOS2 mRNA expression in AECII or AM appeared to depend on the presence of both types of cells together with IFN‐γ.

Time course of reversetranscriptase polymerase chain reaction (RTPCR) for nitric oxide synthase‐2 (NOS2) messenger ribonucleic acid (mRNA) with samples of ribonucleic acid (RNA) isolated from five alveolar macrophages (AM)alveolar epithelial cells type‐2 (AECII) cocultures. AECII with 10% of AM were cultured without (▴) or with tumour necrosis factor‐α (♦), interferon‐γ (▾), interleukin‐1β (▪), or lipopolysaccharide (•) for 0, 4, 8, 16 and 24 h. The PCR products from each culture were individually quantified, and data are presented as the mean percentages of β‐actin density±sem (n=5). **: p<0.01 as compared with unstimulated cells.
In separate experiments, highly purified AECII were cocultured with autologous AM at different percentages (2.5, 5, 10 and 15%) either in the presence or absence 50 U·mL−1 of IFN‐γ for 24 h. As shown in figures 4a and 4b⇓, the IFN‐γ‐induced NOS2 mRNA expression in AMAECII cocultures was dependant on the presence of AM. Levels as low as 5–15% of AM were sufficient to detect their NOS2 mRNA after IFN‐γ stimulation, and further increasing the percentage of AM in AMAECII cocultures did not lead to higher NOS2 mRNA expression levels (fig. 4⇓). As shown earlier, the presence of 5–10% of AECII in the autologous AM population was not enough to induce NOS2 mRNA expression in IFN‐γ‐stimulated cultures (fig. 1⇑ and 2b⇑). As expected, the levels of CD14 mRNA in unstimulated cocultures increased with increasing percentage of AM, whereas the addition of IFN‐γ markedly reduced its accumulation in the AMAECII cocultures (fig. 4⇓). This is in accordance with the observations by Landmann et al. 20 and Payne et al. 21 that IFN‐γ reduces CD14 protein expression in human monocytes. Highly purified AECII preparations in these experiments were free of CD14 mRNA (fig. 1d⇑) and, to the best of the authors' knowledge, no CD14 mRNA or protein expression in human AECII has been described. No changes in SP‐A mRNA were detected (data not shown).

Nitric oxide synthase‐2 (NOS2) and CD14 messenger ribonucleic acid (mRNA) expression in alveolar epithelial cells type‐II (AECII) cocultured with autologous alveolar macrophages (AM) (2.5–15%) in the presence or absence of 50 U·mL−1 interferon‐γ for 24 h. a) Representative gel images from one of two identical experiments. b) Densitometric analysis of gel bands shown in a (□: CD14; ▪: NOS2). Lanes 0 and 11, freshly isolated AECII and AM respectively; lanes 1–10, unstimulated and interferon (IFN)‐γ stimulated AECII with AM; lanes 11, 12 and 13, unstimulated and IFN‐γ‐stimulated AM respectively. M: molecular marker.
Immunocytochemistry and in situ hybridization
A549 cells stimulated with IFN‐γ or a cytokine mixture expressed clearcut NOS2 immunoreactivity as compared to unstimulated cells (fig. 5a and b⇓). In contrast, a weak immunoreactivity for human NOS2 was seen in a small proportion of noncultured AM and it decreased after stimulation with IFN‐γ or with a cytokine combination for 18 h (fig. 5c and 5d⇓). No NOS2 immunoreactivity was detected in unstimulated or cytokinestimulated AECII, which were cultured alone or with 10–15% of autologous AM (fig. 5e and f⇓). Immunostaining of BALAM resulted in a moderate immunoreactivity seen in 10–25% of noncultured cells (fig. 5g⇓). Culture of these cells in the presence of IFN‐γ or a cytokine combination resulted in the same NOS2 staining pattern as was seen in the cells cultured without cytokines for 18 h. In most BALAM preparations a decreasing immunoreactivity was seen after 18‐h culture (fig. 5g and 5h⇓). Slides labelled with buffer or control Ig‐G did not show immunostaining in both AM and A549 cultures (data not shown). In situ hybridization using Diglabelled DNA probes detected specific signals for NOS2 mRNA mainly in intraalveolar macrophages in all lung tissue preparations included in the present report. AM were identified by their size and localization in the lumen of the alveoli in close proximity to alveolar epithelium. In contrast, no positive signals for NOS2 mRNA were detected in alveolar epithelium, including AECII, which were typically localized at alveolar corners (fig. 5i and 5j⇓). Specific signals were not detected in control preparations, in which specific DNA probes were substituted by hybridization buffer alone, or treated with RNase before hybridization (not shown).

Immunocytochemistry for nitric oxide synthase‐2 (NOS2) a) A549 cells unstimulated and b) stimulated with a combination of tumour necrosis factor‐α , interferon‐γ, and interleukin (IL)‐1β for 18 h. c) Alveolar macrophages (AM) freshly isolated from lung tissue samples and d) stimulated with the previously mentioned combination of cytokines for 18 h. e) Alveolar epithelial cells type‐II (AECII) alone and f) with 15% of autologous AM stimulated with the previously mentioned cytokine combination for 18 h. g) Bronchoalveolar lavage cells freshly isolated and h) stimulated with the combination of cytokines for 18 h. Control slides stained without primary antibodies were always negative (not shown). i) and j) In situ hybridization for NOS2 messenger ribonucleic acid (mRNA).
Western blot analysis
Western blot analysis of crude cell lysates of AECII, AM, or AM/AECII cocultures revealed no positive NOS2 protein signals. After enrichment of the cell lysates by 2′5′ADPagarose, NOS2 protein could be detected in IFN‐γ‐stimulated AMAECII cocultures containing 10% AM (fig. 6a⇓). Again, no NOS2 protein was found in lysates from unstimulated cocultures or in either unstimulated or IFN‐γ‐stimulated AECII, whereas an equivalent amount of lysates from LPS/IFN‐γ‐stimulated murine cell line RAW264.7 demonstrated positive signal of protein (fig. 6a⇓). These NOS2 positive control lysates were provided by Transduction Laboratories (Lexington, KY, USA).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
a) Western blot analysis of nitric oxide synthase‐2 (NOS2) production. No NOS2 protein was detected in the cell lysates from unstimulated (unst.), interferon (IFN)‐γ‐stimulated (50 U·mL−1) alveolar epithelial cells (AEC)‐II cultures, and unstimulated alveolar macrophages (AM)‐AEC‐II co‐cultures after 24 h, whereas positive control lysate (C) and lysates from IFN‐γ‐ stimulated AM‐AEC‐II co‐cultures containing 10% AM show clear signals of NOS2 protein. b) In supernatants from AEC‐II cultures minor amounts of total NOx were detectable and in addition 5–15% of AM increased this overall NOx content only marginally, whereas purified AM generated considerably more NOx, and stimulation with IFN‐γ does not significantly influence the generation of NOx in all cases. Bars represent means of duplicate determinations from one representative experiment out of three.
Nitrate/nitrite in alveolar epithelial cells type II/alveolar macrophages supernatants
Supernatants from purified AM contained more NOx compared to supernatants from purified AECII (fig. 6b⇑). Stimulation of purified AM with IFN‐γ did not significantly alter the NOx concentrations in the supernatants. In the AMAECII coculture supernatants only a marginal increase in the overall NOx content compared to AECII cultures could be detected (fig. 6b⇑).
Nitric oxide synthase‐2 messenger ribonucleic acid expression in cells retrieved from bronchoalveolar lavage samples
To exclude any effect of the isolation procedure on NOS2 mRNA expression by human AM from the lung tissue samples, experiments with BALAM obtained from subjects retrospectively free of any systemic or lung diseases were performed. These cells were cultured under the same experimental conditions as AECII and AM from the lung tissue samples. However, no NOS2 mRNA could be found in noncultured, unstimulated or IFN‐γ‐stimulated BALAM. In addition, neither LPS (1 or 10 µg·mL−1) nor a mixture of proinflammatory cytokines, or a combination of both resulted in the induction of NOS2 mRNA (data not shown). However, RTPCR with specific primers for NOS2 yielded the expected band of 462 bp from NOS2 positive controls (IFN‐γ‐stimulated A549 cells), and all RNA samples were positive for β‐actin mRNA (data not shown).
In order to evaluate whether SP‐A might be the AECIIderived factor able to enhance IFN‐γ‐induced cell responses, SP‐A at different concentrations (1–100 µg·mL−1) was added in the presence or absence of IFN‐γ to BALAM. Neither SP‐A alone nor a combination of IFN‐γ and SP‐A at different concentrations resulted in the induction of NOS2 mRNA in BALAM (data not shown).
Discussion
There is growing evidence that certain forms of acute and chronic pulmonary inflammation such as bronchial asthma 22, ARDS 5, IPF 3, 9, tuberculosis 1, and sarcoidosis 2 are associated with the upregulation of pulmonary NOS2 expression. In addition, an increase of NO in exhaled air is measured in these diseases. Although the sources of NO under these pathophysiological conditions are not completely clarified, it is generally thought that AM and airway epithelium represent the major sources.
Human AM contain NOS2 protein and can express the NOS2 gene. The accumulation of NOS2 protein has been documented in AM isolated from patients with active pulmonary tuberculosis 8, lung cancer 4, and ARDS 3. In addition, it has been shown that AM in lung sections from patients with IPF 9, chronic obstructive pulmonary disease (COPD) 23 and sarcoidosis 2 display strong immunoreactivity for NOS2 protein in situ. However, many attempts to induce NOS2 mRNA expression and/or NO production by human AM in vitro failed 7, 24, 25. As shown in the present investigation, AM isolated from lung tissue samples exhibited immunoreactive NOS2 protein ex vivo and its level was preserved or slightly decreased during 18‐h culture periods with or without stimuli such as proinflammatory cytokines. In addition, AM, but not AECII, were strongly positive for NOS2 mRNA in situ on lung tissue preparations included in this report. However, neither lung tissue derived nor BAL retrieved human AM express detectable NOS2 mRNA levels in the absence or presence of proinflammatory cytokines or LPS in vitro. Interestingly, these findings corroborate with previously published results, stating that AM retrieved from BAL cells of normal donors and patients with pulmonary tuberculosis or primary lung cancer contained NOS2 protein ex vivo, but no NOS2 mRNA 4, 7, 8, 24. This was true for cells, which were stimulated by IFN‐γ, LPS or mycobacteria in vitro 8. It can be concluded that the alveolar microenvironment is a critical factor for accumulation and/or stability of NOS2 mRNA in human AM.
There are many factors including direct cellcell contacts and soluble mediators that influence NOS2 expression of human AM. A recent study indicates that human SP‐A enhances production of NO and NOS2 protein expression in rat AM stimulated with IFN‐γ or with LPS/IFN‐γ combinations. SP‐A itself did not affect production of NO or NOS2 protein expression in rat AM 12. In contrast to rat AM, the present results demonstrate that neither IFN‐γ alone nor the combination of IFN‐γ with SP‐A induced NOS2 gene transcription in human BALAM in monoculture. However, these cells, as well as AM isolated from lung tissue samples, contained immunoreactive NOS2 protein and its expression levels decreased after 18‐h culture with or without proinflammatory cytokinestimulation. These unexpected differences between NOS2 mRNA and protein expression levels in human AM can be explained by a very low stability of NOS2 mRNA in combination with a long halflife time of the protein. Regulatory processes acting on NOS2 mRNA stability and protein halflife have been identified in human articular chondrocytes and mouse peritoneal macrophages 26, 27.
The requirements for the induction of NOS2 gene transcription in human monocytes/macrophages depend on the origin of the cells 7, 28. Using protocols, which elicit NOS2 mRNA expression in peripheral blood derived macrophages 28 NOS2 mRNA in human AM irrespective of source and in vitro stimulation could not be detected (fig. 2b⇑). The authors hypothesized that NOS2 mRNA expression and its steadystate levels in human AM depend on in situ conditions within the alveolar space, and this may explain the conflicting results of NOS2 mRNA and protein expression in vivo and in vitro. Following these observations the authors were stimulated to evaluate whether the presence of human AECII is the factor inducing NOS2 expression in human AM.
The in vivo stimulus for NOS2 expression and NO production by human AM has not been elucidated. However, the close proximity of these cells and AECII within the alveoli suggests that AECderived products as well as direct cellcell contacts might be involved. The present results demonstrate that human AM and AECII, when stimulated separately by IFN‐γ do not express NOS2 mRNA within a 24‐h culture period, whereas the coculture of these cells in the presence of IFN‐γ is the most potent factor for the induction and steadystate level of NOS2 mRNA. These findings indicate that NOS2 mRNA can be expressed in IFN‐γ‐stimulated AMAECII cocultures at a percentage of AM as low as 2.5%, and the level of NOS2 mRNA increased with the increasing percentage of AM in AMAECII cocultures up to 10% of cells. Further increase of the AMAECII ratio did not lead to an increase in NOS2 mRNA accumulation. AM obtained from lung tissue were contaminated with 5–10% of AECII as indicated in the Results section (fig. 1c and 1d⇑). However, despite the presence of AECII in these AM preparations, neither unstimulated nor IFN‐γ‐stimulated cells express NOS2 mRNA (fig. 2b and 4⇑⇑). Accordingly with NOS2 mRNA data, the levels of NOS2 protein and NOx concentrations also increased with the increasing of AM percentage in the IFN‐γ‐treated cocultures. Although there were no differences between total NOx concentrations in supernatants from unstimulated and IFN‐γ‐stimulated AMAECII cocultures, a small amount of NOS2 protein was only detected in cell lysates from the latter (fig. 6a⇑). In addition, immunostaining of AECII and AM in coculture with antibodies against human NOS2 showed that immunoreactive protein was expressed in AM, but not in AECII as identified by cell shape and size (fig. 5c and 5f⇑). It is worth noting that NOS2 immunoreactivity was decreased in cytokinestimulated AM in comparison with noncultured cells. These observations may explain the absence of differences in NOx generation, because total amount of NOS proteins and their enzymatic activities are too low for generating NO quantities detected by such an indirect method as the Griess reaction.
The authors have shown that neither NOS2 mRNA nor protein could be detected in human AECII in vivo, ex vivo or upon cytokinestimulating conditions in vitro. However, the cell line A549, which was used as the control, expressed NOS2 mRNA and protein upon IFN‐γ‐ or TNF‐α‐stimulation. These data corroborate well the results of Robbins et al. 29. In contrast to the current study, others have demonstrated a stimulatory influence of LPS and proinflammatory cytokines, including IFN‐γ on NO production and NOS2 expression by rat AECII 30, 31. The reason why the stimuli mentioned earlier appeared to induce AM and AECII in a rat model, but did not in humans is not clear. There is some evidence that species differences may significantly affect the outcome of experiments for NOS2 induction in AM and AECII 32, 33. It may also be possible that the stimuli and culture conditions, which were used here, are inadequate to stimulate human AM and AECII to express NOS2 mRNA and, therefore, do not simulate the complex dynamic processes occurring in vivo, especially during a lung inflammation. In this respect Saleh et al. 9 have demonstrated a weak expression of nitrotyrosine and NOS2 in the airway epithelium and AM of normal lung tissues, which were obtained from the normal part of human lungs by pneumectomy due to lung cancer. Furthermore, it has been shown that in lung cancer patients the production of NO from AM was increased as a result of the in vivo upregulation of NOS2 activity, and not associated with tumour localization in the lungs 4. The in vitro and in situ hybridization data in this study corroborate well with both observations cited earlier. Thus, all these findings suggest that the NOS2 mRNA accumulation and protein production in AMAECII coculture is attributable to AM, and both the AMAECII ratio and the presence of IFN‐γ are critical factors for the NOS2 mRNA induction in human AM under experimental conditions tested.
From the present study, it is not yet clear how AECII mediate the upregulation of NOS2 expression by AM. Possible mechanisms include the production of mediator(s) or direct celltocell interactions with involvement of a critical surface expression pattern of known adhesion molecules, or altering the sensitivity of the AM to external cytokine stimulation including IFN‐γ and IL‐1β. These questions are currently under investigation.
To conclude, nitric oxide synthase‐2 is the enzyme responsible for the generation of large amounts of nitric oxide, which contribute to inflammatory responses elicited by endotoxin, cytokines or physicochemical stress. In vitro, human alveolar macrophages have repeatedly been shown to be hyporesponsive in nitric oxide synthase‐2 expression and nitric oxide production in response to inflammatory stimuli as compared with monocytes/macrophages isolated from peripheral blood 7, 28. It is likely that, nitric oxide synthase‐2 expression and nitric oxide generation in the alveolar compartment of the human lung are under a tight control to protect the host from nitrous oxide or peroxynitrite radicals, which might cause progressive destruction and/or fibrotic remodelling of lung tissue. The authors propose that alveolar epithelial cells typeII play an important role in this control.
Acknowledgments
The authors would like to thank N. Husmann and S. Adam for technical assistance.
- Received May 15, 2001.
- Accepted November 8, 2001.
- © ERS Journals Ltd
References









