





Abstract
It is unclear whether inflammation in the cystic fibrosis (CF) lung relates predominantly to bacterial infection, or occurs as a direct consequence of mutant cystic fibrosis transmembrane conductance regulator (CFTR) protein.
Interleukin (IL)-8 secretion from CF and non-CF cell lines, and from CF and non-CF human primary nasal epithelial cells incubated with or without Pseudomonas aeruginosa, was measured. Activation of nuclear factor-κB (NF-κB) in unstimulated CF and non-CF nasal epithelial cells, cell lines and murine tissues was measured by gel-shift assays.
No significant difference in basal IL-8 production or NF-κB activation was observed between CF and non-CF primary nasal cells. However, CF cells exhibited a significantly (p<0.01) increased IL-8 secretion following P. aeruginosa stimulation. Equalization of the increased P. aeruginosa adherence observed in CF cells, to non-CF levels, resulted in comparable IL-8 secretion. Further, IL-8 production did not differ with mutations which result in either correctly localized CFTR, or in partial/total mislocalization of this protein. Similar levels of NF-κB activation were observed in a number of organs of wild-type and CF mice. Finally, IL-8 secretion and NF-κB activity were not consistently increased in CF cell lines. Cos-7 cell transfection with plasmids expressing ΔF508 or G551D mutant CFTR protein resulted in increased activation of a p50-containing NF-κB complex, but IL-8 secretion was similar to wild-type cells.
The authors conclude that the stimulus produced by Pseudomonas aeruginosa is the predominant inflammatory trigger in their models.
- cystic fibrosis trans-membrane conductance regulator
- endoplasmic reticulum
- interleukin-8
- nuclear factor-κB
- Pseudomonas aeruginosa
This study was supported by the Association Française de Lutte contre la Mucoviscidose, the Société de Pneumologie de Langue Française, the European Respiratory Society, the North Atlantic Treaty Organization, the Cystic Fibrosis Research Trust and a Wellcome Trust Senior Clinical Fellowship.
Over the last two decades, the life expectancy of patients with cystic fibrosis (CF) has improved dramatically, allowing median survival to reach thirty years. Cloning of the CF gene and subsequent investigations into the molecular physiology of the gene product have led to a better understanding of the disease. The CF gene encodes the cystic fibrosis transmembrane conductance regulator protein (CFTR), a chloride channel localized to the apical surface of epithelial cell membranes. More than 800 CF mutations are classified into five groups based on the mechanism by which the function of CFTR is disrupted. Broadly, these include those in which no protein is produced (class I), protein which does not reach its correct location (class II), and those in which smaller amounts or less functional protein are correctly localized (classes III–V) 1. The deletion of the amino acid phenylalanine at position 508 (ΔF508) in the CFTR protein accounts for >70% of CF alleles in Caucasians and is an example of a class II mutation.
The most common cause of death in CF is respiratory failure as a result of chronic pulmonary infection and inflammation 2. However, it is not clear whether the inflammation is simply a secondary response to infection, or whether the primary defect in CFTR is more directly linked to the generation of this inflammatory process. A number of lines of evidence link mutant CFTR with an increased susceptibility to infection. Most studies suggest that CF subjects show reduced mucociliary clearance, perhaps related to the primary defect. This, in turn, will predispose to infection by a variety of organisms 2. Local host antimicrobial defences, such as β‐defensins 3, may show impaired function as a result of alterations in airway surface fluid (ASF) electrolytes, although it is presently unclear whether CF ASF is indeed abnormal with regard to sodium or chloride content 4. A controversial hypothesis suggests that there is a defect in ingestion of Pseudomonas aeruginosa by respiratory epithelial cells 5, likely to facilitate infection. Finally, the receptor for P. aeruginosa, the asialoglycolipid asialo-GM1, is significantly increased on the apical cell membrane of CF respiratory epithelium and may, therefore, promote infection by increasing adherence 6. Thus, several studies support the hypothesis that mutant CFTR can lead to increased bacterial colonization and resultant inflammation.
The hypothesis of primary (endogenous) inflammation is based firstly on clinical observations of inflammation preceding detectable infection in CF neonates and young children. Thus, bronchoalveolar lavage fluid showed neutrophil infiltration, increased levels of interleukin (IL)-8 7, 8 and a protease/antiprotease imbalance, but was negative for bacterial culture 7. A recent study adds weight to the concept of endogenous inflammation 9. Early inflammation was examined in human foetal airway xenografts in severe combined immunodeficient (SCID) mice. Significant increases in IL‐8 secretion were seen from CF proximal airway grafts prior to infection, when compared to non-CF grafts. Neutrophil recruitment to the basal membrane and movement to the epithelial surface was also evident. This was slower and less marked in non-CF proximal airways. In distal airway xenografts, neutrophil migration was again observed in CF airways, but was not seen in non-CF tissues. One possible explanation for endogenous inflammation is that mutant (mistrafficked) CFTR protein may directly increase activation of the transcription factor nuclear factor-κB (NF‐κB) 10. NF‐κB, and particularly the p50/p65 heterodimer, is involved in the regulation of a large number of proinflammatory mediators 11. In addition, “protective” cytokines such as IL‐10 which serve to terminate inflammation, and are also under the control of NF‐κB, have been shown to be significantly reduced in CF 12.
IL-8 is a powerful chemoattractant and is transcriptionally regulated by NF-κB. IL-8 is involved in recruitment of neutrophils into the lung 13 or nasal mucosa 14 and is a central factor in the inflammatory response of the airways 15. This study has examined whether there is evidence for basally increased IL‐8 production and NF-κB activation to support the hypothesis directly linking mutant CFTR to inflammation. To assess the alternative hypothesis of inflammation following infection, it has been studied whether in CF, stimulation with P. aeruginosa results in exaggerated production of IL-8, and whether this in turn relates to the characteristically increased adherence level of these organisms.
Materials and methods
Patient population
The study used 29 adult CF patients (21 males) with a mean (range) age of 29.2 (17–44) yrs. None were using nasally inhaled, oral or intravenous steroids and no patient was studied during an acute exacerbation. The genotype of each subject is included in the Results section. Eighteen nonsmoking healthy volunteers (11 males) with a mean age of 32.7 (23–50) yrs served as controls. The protocol was approved by the Royal Brompton Hospital Ethics Committee and all subjects gave informed consent.
Nasal epithelial cell harvesting
Nasal epithelial cells were obtained by brushing the mucosa of the inferior turbinate of both nostrils with a 3 mm cytology brush (Diagmed, Thirsk, UK). Recovered cells were suspended in Ham's F12 medium (Gibco BRL, Paisley, UK), washed once and pelleted by pulse centrifugation (13,000×g) in phosphate-buffered saline (PBS) and resuspended in Ham's F12. Cell numbers were quantified by microscopic examination in a standard haemocytometer counting chamber (Sigma, Poole, UK) and viability evaluated using the trypan blue exclusion assay. Subsequently, samples were divided into two groups for assessment of IL‐8 secretion and bacterial adherence. Viability was again quantified after 24 h.
Bacterial strain
A laboratory reference strain of P. aeruginosa (international antigenic typing scheme (IATS) serotype 0:1 (National Collection of Type Cultures 11440, American Type Culture Collection 3348), nonmucoid, piliated and gentamycin-sensitive) was used 16. The bacteria were grown on blood agar plates for four consecutive days before overnight culture in tryptone soya broth (Unipath Ltd, Basingstoke, UK) to ensure piliation. Piliation status was confirmed by transmission electron microscopy. Briefly, bacteria were suspended in distilled water, placed onto formvar coated grids (Agar Scientific Ltd, Stansted, UK) and negatively stained with 0.65% sodium phosphotungstate. Subsequently, bacteria were pelleted by centrifugation at 160×g for 10 min, supernatant was removed and the pellet resuspended in 1 mL of phosphate buffered saline (PBS). Bacterial concentration was quantified by spectrophotometry, with an optical density at 620 nanometers, of 0.1 corresponding to ∼2.5×108 bacteria, as previously described 17. Bacterial dilutions were prepared in PBS.
Pseudomonas stimulation assay
Nasal epithelial cells were divided into two 400-µL aliquots. P. aeruginosa at a final concentration of ∼5.2×107 bacterial·mL−1 was added to one aliquot, whilst the second aliquot was used to determine basal IL-8 secretion in the absence of stimulation. Both aliquots were incubated at 37°C with 5% carbon dioxide. After 1 h, 50 µL of supernatant were removed from each sample and immediately stored at −20°C for subsequent IL-8 analysis. Gentamycin (500 µg·mL−1) was added to each sample and the incubation was continued under the same conditions. After 24 h, 50 µL of supernatant were removed from each aliquot and stored at −20°C. IL‐8 quantification was carried out using a standard enzyme-linked immunosorbent assay (ELISA; Genzyme, Billinghurst, UK), with results expressed as IL-8 production·10−5 live cells present at the start of the incubation.
Assessment of bacterial adherence by scanning electron microscopy
Adherence was assessed by a previously described technique 17. Briefly, after incubation at 37°C for 1 h, nasal brushings were spun through a 50% Percoll gradient (Pharmacia-Biotech, St Albans, UK) at 4°C at 13,000×g for 20 min, to separate the nonadherent bacteria from bacteria attached to epithelial cells. The nonadherent bacteria pelleted at the base of the tube, whereas bacteria attached to epithelial cells remained at the percoll/PBS interface. This fraction was removed and cytospun at 20×g onto a thermanox coverslip (Emitech, Ashford, Kent, UK). Cells were fixed overnight with 2.5% cacodylate-buffered glutaraldehyde, washed in sodium cacodylate buffer and dehydrated with serial concentrations of ethanol and hexamethyldisilazane (TAAB Laboratories, Aldermaston, UK). Coverslips were then mounted onto aluminium stubs, coated with gold, and analysed through field-emission scanning electron microscopy (Hitachi S4000, Nissei Sangyo, Tokyo, Japan). Samples were coded and examined with the observer blinded to the incubation conditions. Bacterial binding, defined as direct contact of P. aeruginosa with the ciliated surface of the cells, was assessed in a mean±sem of 41±3 cells per sample. Adherence data are expressed as the mean±sem of adherent bacteria per 10 cells.
Equalization of Pseudomonas aeruginosa adherence
As increased IL‐8 secretion levels produced by CF cells may simply reflect the increased receptor number, the authors attempted to equalize bacterial adherence in CF and non-CF cells. Decreasing dilutions of the P. aeruginosa standard culture were applied to CF cells, until similar adherence levels were seen in CF and non-CF cells.
Electrophoretic mobility shift assay
Cell pellets were washed twice in ice-cold PBS (Sigma, Poole, UK) and lysed in 70 µL of extraction buffer as previously described by Pahl et al. 18. The authors assayed total cellular protein, since irrespective of intracellular localization, only activated NF‐κB can form deoxyribonucleic acid (DNA)/protein complexes, and thus be visualized using the electrophonetic mobility shift assay (EMSA). Briefly, cells were resuspended in extraction buffer (20 mM N-[2-hydroxyethyl] piperazine-N’-[2-ethanesulfonic acid] (HEPES), pH 7.9, 350 mM NaCl, 20% glycerol, 1% Nonidet (NP40), 0.5 mM ethylenediamine tetraacetic acid (EDTA), 1 mM ethyleneglycol-bis-(β‐aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA), 0.5 mM dithiothreitol (DTT) plus protease inhibitors (0.2 trypsin inhibitor units (TIU) mL−1 aprotinin, 500 µM 4-(2-aminoethyl)benzinesulphonyl fluoride (AEBSF), 1 mM sodium orthovanadate, 10 mM sodium fluoride) and incubated on ice for 30 min. Cell debris was removed through centrifugation (14,000×g 5 min) and the protein concentration was determined by a modified Folin-Lowry method 19. The gel shift assay system from Promega (Southampton, UK) was used and EMSA was carried out according to manufacturers recommendations. An oligonucleotide probe containing the κ light chain enhancer consensus sequence binding site for NF‐κB (5′-AGT TGA GGG GAC TTT CCC AGG C-3′) was end-labelled with radiolabelled adenosine triphosphate (32P-ATP) (Amersham Life Science Ltd, Little Chalfont, UK). Each reaction used 10 µg of cellular protein. Reaction products were resolved on 5% nondenaturing polyacrylamide gels (Anachem, Luton, UK) at 10 mA for 3 h at 20°C. Gels were then dried for 2 h at 80°C and exposed to Kodak Biomax MS film at −80°C. To identify the specificity of NF‐κB binding, competition studies with an approximate 50-fold excess of unlabelled NF‐κB consensus oligonucleotide were carried out. For supershift analysis, 2 µg of either anti-p50, anti-p52, anti-cRel, anti-RelB or anti-p65 antibodies (Santa Cruz Biotechnology Inc., CA, USA) were added after the addition of labelled oligonucleotide and reactions were incubated for a further 45 min at room temperature. Quantification of NF‐κB complexes was carried out by densitometry using LabScan and ImageMaster 1D software (Amersham Pharmacia Biotech, St. Albans, UK).
Mice
Lungs, trachea, liver, spleen and kidneys were removed from ΔF508 homozygote CF mice (CFTRtm2Cam) 20 and non-CF littermates (age 6–12 weeks). To determine NF‐κB activity the tissues were mechanically homogenized in 1 mL of extraction buffer and processed for EMSA as described above. Each reaction used 20 µg total protein.
Cell culture
Human CF cell lines used were tracheal epithelium (ΣCFTE29o-, ΔF508/ΔF508), submucosal epithelium (CFSMEo-, ΔF508/unknown) and nasal polyp (CF-NPE9o-, unknown/unknown). Human non-CF cell lines used were bronchial epithelium (16HBE14o-), tracheal epithelium (9HTEo-) and tracheo-bronchial epithelium (TBEo-). All cell lines were provided by Dieter Gruenert (University of California, San Francisco, CA, USA). CFNP9o-, ΣCFTE29o-, CFSMEo- and 16HBE14o- cells were cultured in minimal essential medium (MEM); 9HTEo- and TBEo- cells were grown in Dulbeccos modified Eagle medium (DMEM). Both media were supplemented with 10% foetal calf serum (FCS) and 1% penicillin-streptomycin solution and cells grown in a 5% CO2 atmosphere. All media, FCS and antibiotic solutions were obtained from Sigma (Poole, UK). Cultures were assayed for mycoplasma contamination using an ELISA assay (Boehringer Mannheim UK, Lewes, UK) and were shown to be free of infection. IL‐8 secretion during 24 h incubation was determined in semiconfluent monolayers using a standard ELISA assay. To determine NF‐κB activation cells were grown to 70% confluence, scraped into surrounding medium and pelleted at 160×g for 5 min at room temperature and protein lysates were prepared as described above. The degree of cell confluence in immortalized cell culture may influence activation of NF‐κB proteins. Thus, care was taken to ensure that all cell cultures were assayed at 70% confluence. Cos‐7 cells (supplied by ATCC) were cultured under routine procedures in DMEM supplemented with 10% FCS and 1% streptomycin/penicillin.
Transfections
The 6.2 kb wild-type, ΔF508 and G551D CFTR complementary deoxyribonucleic acids (cDNAs) were cloned into the Not I site of the eukaryotic expression vector pCMVβ (Clontech Laboratories UK Ltd, Basingstoke, UK), from which the β‐galactosidase reporter gene was removed through Not I digestion. DNA was prepared using Qiagen columns (Qiagen Ltd, West Sussex, UK), according to manufacturers recommendations. Transient transfection of semiconfluent Cos‐7 cells was carried out using plasmids complexed to the cationic lipid DC-Cholesterol/dioleylphosphatidylethanolamine (DOPE) at a ratio of 1:3 (44.8 µg plasmid: 134.4 µg lipid per 80 cm2 flask) in Optimem (Sigma). The medium was replaced with normal growth medium 24 h after transfection. IL‐8 was determined 48 h after transfection and NF‐κB activity was determined as described above.
Statistical analysis
The Mann-Whitney U‐test was used to compare means and the Wilcoxon rank sum test to assess significance of changes from background levels. The null hypothesis was rejected at p<0.05. Data are presented as mean±sem for convenience. Numbers of human subjects or mice are indicated by n numbers, where appropriate.
Results
Interleukin-8 secretion by nasal epithelial cells
In the absence of added P. aeruginosa, IL‐8 secretion at 24 h from freshly obtained CF (n=19) and non-CF (n=16) primary nasal epithelium was not significantly different (fig. 1⇓). Incubation with P. aeruginosa resulted in a significant (p<0.05) increase in IL-8 secretion after 24 h from CF, but not from non-CF nasal cells. Thus, in the presence of P. aeruginosa, IL‐8 secretion from CF cells was approximately three-fold higher than from non-CF cells (p<0.01). Assessment of cell viability by trypan blue exclusion and evidence of ciliary beating demonstrated that there was no significant difference in the number of live cells at 24 h after stimulation (CF: 1.8±0.6×105·mL−1, n=11; non-CF: 1.7±0.5×105·mL-1, n=8).

Interleukin-8 secretion assayed over 24 h from nasal epithelial cells under basal conditions and following exposure to Pseudomonas aeruginosa (cystic fibrosis (CF) n=19, non-CF n=16). Error bars indicate sem. □: without P. aeruginosa; └: with P. aeruginosa; *: p<0.05 versus unstimulated CF cells; **: p<0.01 versus stimulated CF cells.
Equalization of Pseudomonas aeruginosa adherence levels in cystic fibrosis and non-cystic fibrosis cells
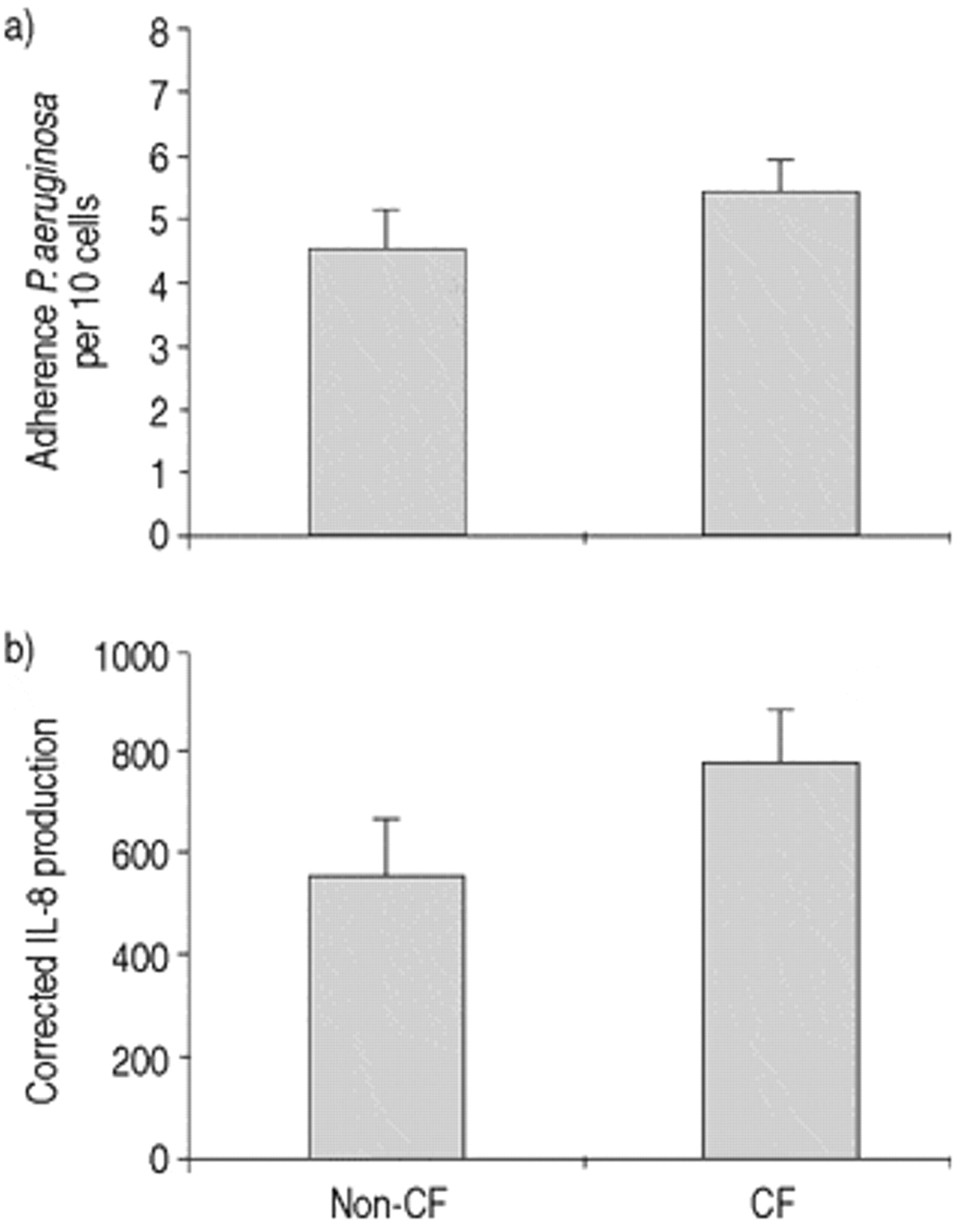
It has previously been demonstrated using these methods that P. aeruginosa adherence to CF cells is ∼3–4 times greater than to non-CF cells 17. As this may have been the stimulus for IL-8 production, the authors attempted to equalize the numbers of adherent bacteria to CF and non-CF cells. The authors first assessed the highest concentration of P. aeruginosa which could be applied to non-CF cells without affecting their viability, as assessed by trypan blue exclusion and evidence of ciliary beating. A 1:4 dilution of the standard P. aeruginosa culture, corresponding to 1.6×108 bacteria·mL−1, was the maximum tolerated concentration resulting in 4.5±0.6 P. aeruginosa adhering per 10 cells (n=7).
Next, the P. aeruginosa concentration applied to CF cells was reduced to produce similar levels of binding to that seen in non-CF cells (fig. 2a⇓). This level of binding was reached for CF cells with a P. aeruginosa solution approximately six times less concentrated than for non-CF, corresponding to 2.5×107 bacteria·mL−1. Viability, assessed at 24 h, using these two concentrations of P. aeruginosa was not different in CF and non-CF cells.

a) Equalization of Pseudomonas aeruginosa adherence between cystic fibrosis (CF) and non-CF cells. Cells were incubated with final concentrations of 1.6×108 and 2.5×107 bacteria·mL−1, respectively. b) Interleukin-8 (IL-8) secretion from the same cells measured 24 h after stimulation (non-CF n=7, CF n=13). Note that the vertical axis represents IL-8 in units of pg·mL−1, corrected for the number of adherent organisms in each individual sample. Error bars indicate sem.
Interleukin-8 secretion under conditions of similar Pseudomonas aeruginosa adherence
When levels of P. aeruginosa adherence were approximately equal, IL‐8 production by CF (n=13) and non-CF cells (n=7) did not differ over a 24 h period (fig. 2⇑). Note that IL‐8 values for this comparison were normalized for the number of adherent bacteria demonstrated for each subject to try to ensure greater accuracy. Because of this correction, values differ in magnitude from those shown in figure 1⇑. As different concentrations of bacteria were applied to CF and non-CF cells, adherence to the basolateral membrane was also examined to rule out any effects this may have on IL‐8 secretion. No significant difference in the degree of basolateral adherence was observed between CF and non-CF cells (CF: 3.0±0.5·10 cells−1; non-CF: 2.0±0.4·10 cells−1).
Influence of genotype
To determine whether CF genotype influences P. aeruginosa induced-IL‐8 secretion, patients were classified into three groups. Group 1 (n=13) with mistrafficking mutations on both alleles (ΔF508 homozygotes and a ΔF508/I507 compound heterozygote), group 2 (n=6) with only one mistrafficking mutation (ΔF508 compound heterozygotes, the second mutation being R553X×2, 1717-1G→A, 621+1G→T, G551D×2), and group 3 (n=3) without mistrafficking mutations (genotypes being G542X/3849+10κB G→T, G542X/R533X, 1717-1G→A/G551D). No difference was found in IL‐8 secretion between the three groups (fig. 3⇓).

Influence of cystic fibrosis genotype on interleukin-8 secretion. Group 1: mistrafficking mutation on both alleles (n=13); Group 2: one mistrafficking mutation (n=6); Group 3: both nonmistrafficking mutations (n=3). Error bars indicate sem.
Nuclear factor‐κB activity in primary nasal epithelial cells and in cystic fibrosis murine tissues
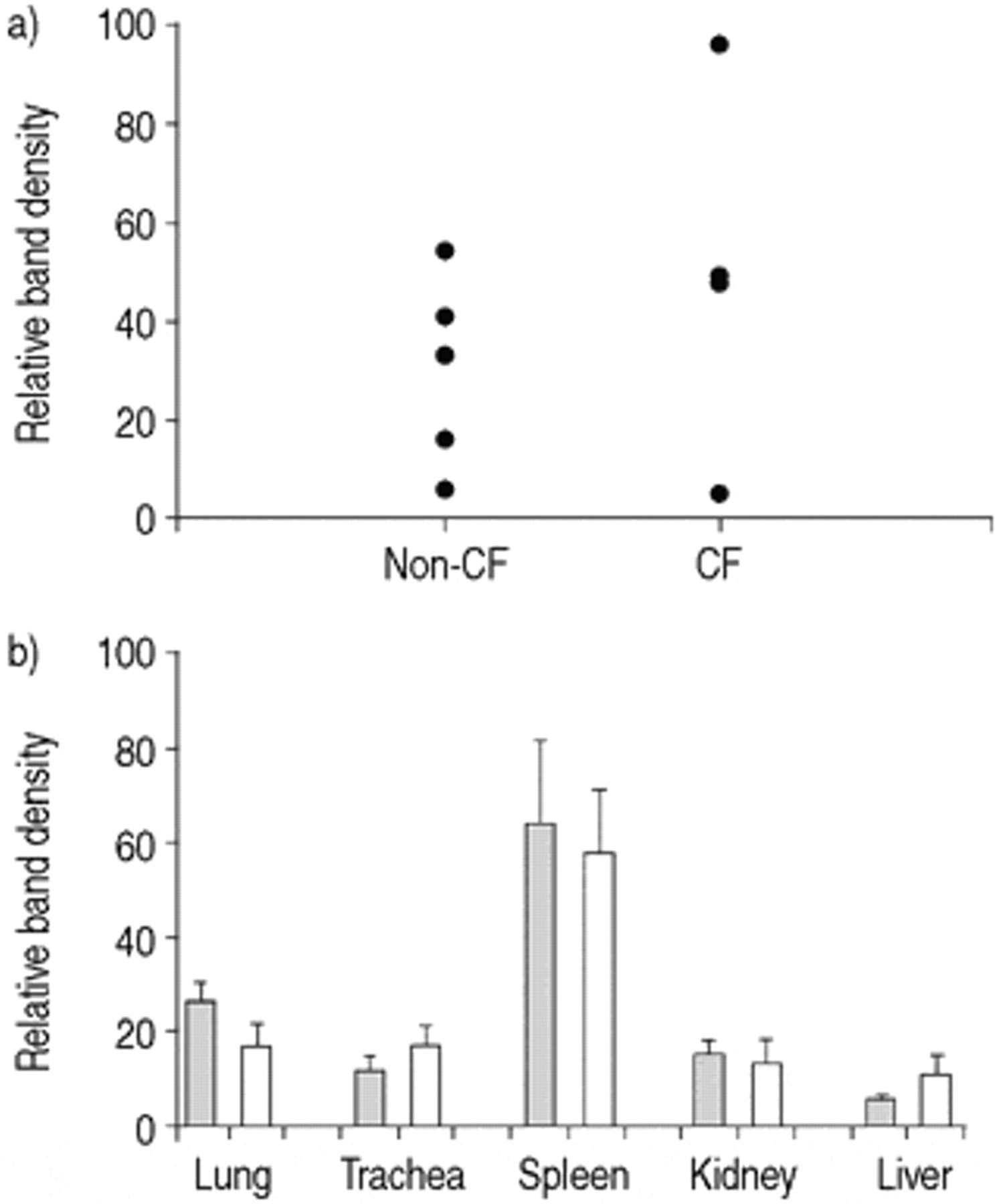
EMSA assays using NF‐κB consensus oligonucleotides resulted in the appearance of three specific DNA/protein complexes in all systems studied. A typical example from unstimulated 9HTEo- cell lysates is shown in figure 4⇓. Only DNA/protein complexes I and II are increased after tumour necrosis factor‐α (TNF‐α) and P. aeruginosa stimulation in cell lines and in mice (unpublished data). Complexes I and II were, therefore, quantified in combination in subsequent experiments as a marker of activated NF‐κB. Supershifts indicated that complex II contains p50 protein, but the composition of complex I could not be determined (fig. 4⇓). To eliminate any variations in complex I and II band density due to individual gel experimental procedures, bands of interest were standardized to the band density of a nonspecific band in each lane. NF‐κB activity was similar in freshly obtained primary nasal epithelial cells derived from CF (n=4) and non-CF subjects (n=5) (fig. 5⇓).

Electrophoretic mobility shift assay (EMSA) using unstimulated 9HTEo- cell lysates. EMSA assays carried out using a nuclear factor-κB (NF-κB) consensus binding site resulted in three specific protein/deoxyribonucleic acid complexes. a) A typical example using lysate from unstimulated 9HTEo- cells. Lanes 1 and 2: Duplicate 10 µg lysates incubated with radiolabelled NF-κB consensus binding site. Complexes I, II (p50 containing) and III are indicated. Lane 3: competition with unlabelled consensus oligonucleotide. *: nonspecific complex. b) Supershift assay using unstimulated 9HTEo- lysates. Lane 1: no added antibodies. Lanes 2–4: addition of antibodies specific to p50 (lane 2), p65 (lane 3) or c-Rel (lane 4) containing components of NF-κB. A shift occurred only with the anti-p50 antibody.

Quantification of total nuclear factor-κB (NF-κB) activity in cystic fibrosis (CF) and non-CF primary nasal epithelial cells and CF and non-CF murine tissues. a) Nasal epithelial cells from non-CF (n=5) and ΔF508 CF subjects (n=4). b) Tissues from wild-type (└) and ΔF508 CF littermates (□). Because separate organs were analysed on individual gels, inter-organ comparison of NF-κB activity cannot be made. Error bars indicate sem.
These observations were extended to a comparison of CF and non-CF murine organs. There was also no significant difference in NF‐κB activation in lung, trachea, spleen, kidney and liver of ΔF508 CF mice (n=6) and wild-type littermates (n=6) (fig. 5⇑). For technical reasons it was not possible to assess the intestinal tract.
Nuclear factor‐κB activity and interleukin‐8 secretion in immortalized cell lines
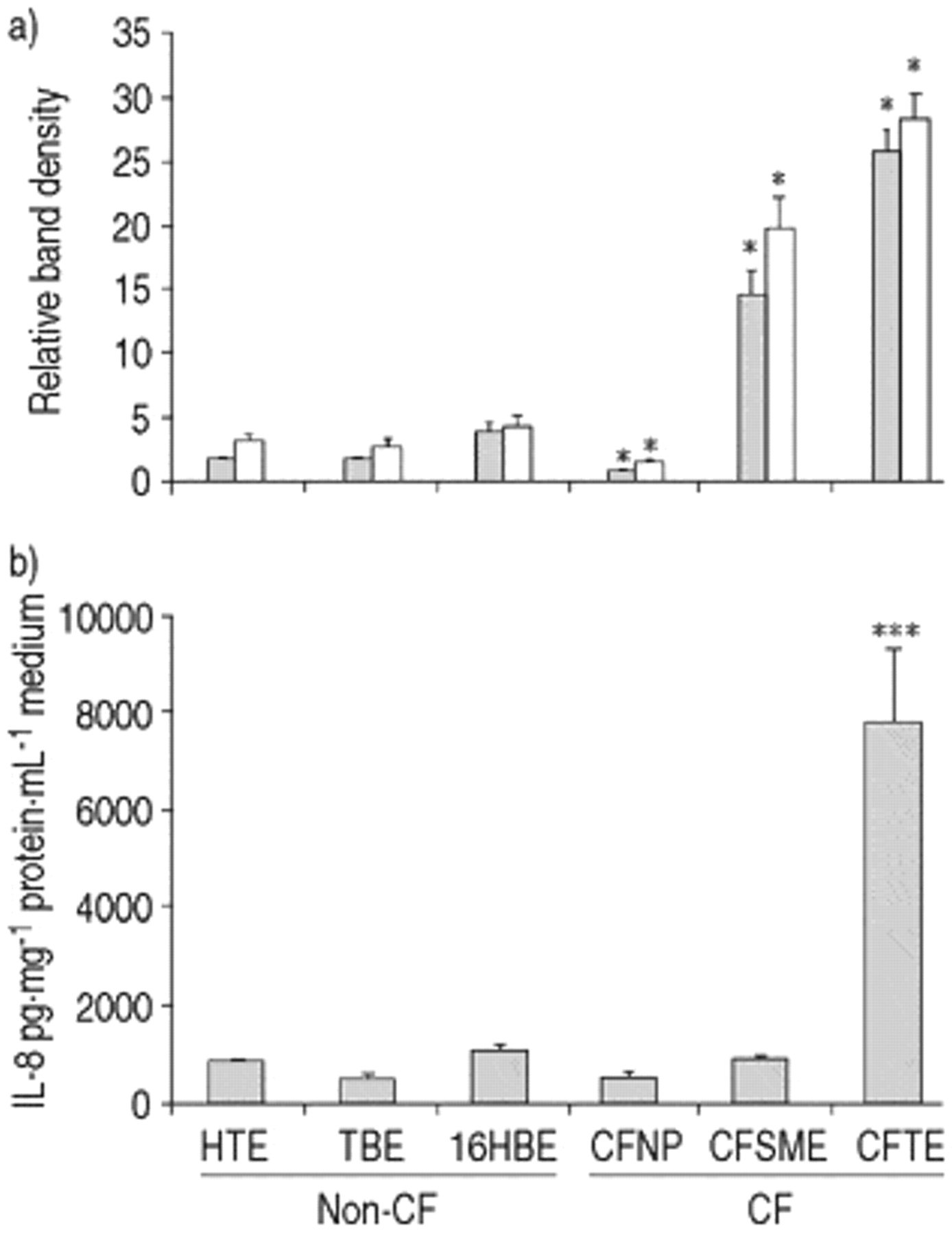
CFNP9o- cells exhibited a significant decrease in activation of NF‐κB (n=6, p<0.001) and no difference in IL‐8 secretion when compared with the mean of the three non-CF cell lines (fig. 6⇓). In CFSMEo- cells NF‐κB activity was increased (n=6, p<0.01), but IL‐8 secretion was not different (n=6). ΣCFTE29o- cells showed an increased activation of NFκB (n=6, p<0.001) and IL‐8 secretion (n=6, p<0.001). The increased NF‐κB activity in CFSMEo- and ΣCFTE29o- cells was largely due to an increase in complex II.

Nuclear factor-κB (NF-κB) activation and interleukin-8 (IL‐8) secretion in unstimulated cystic fibrosis (CF) and non-CF cell lines. Quantification of a) complex II (└), complex I+II (□) and b) IL-8 secretion (n=6, *: p<0.001, versus mean of non-CF values). Error bars indicate sem.
The effects of transfection with wild-type, ΔF508 and G551D cystic fibrosis transmembrane conductance regulator on nuclear factor-κB activity and interleukin‐8 secretion
Cos‐7 cells transfected with either ΔF508 or G551D CFTR plasmids, exhibited a significant increase in p50-containing complex II when compared to wild-type transfectants (n=10, p<0.01) (fig. 7⇓). No significant increase in complex I was observed. IL‐8 secretion was not significantly different in any of the transfectants (fig. 7⇓). Successful transfection was confirmed through plasmid-specific reverse transcriptase polymerase chain reaction (RT-PCR; data not shown). Following transfection with a β-galactosidase reporter gene 11±3% (n=6) of cells were positive for an X‐gal immunohistochemical assay. However, this assay is known to underestimate the overall transfection efficiency 21.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of wild-type and mutant cystic fibrosis transmembrane regulator (CFTR) overexpression on nuclear factor-κB (NF-κB) activity and interleukin-8 (IL-8) secretion. Cos-7 cells were transfected with plasmids expressing wild-type (WT), ΔF508 and G551D CFTR for 48 h. a) NF-κB complexes (□) and II (p50 containing) (└) were quantified and data were expressed as ratios between wild-type and ΔF508 or G551D mutant protein (n=10, *: p<0.01 expressed as significance from a ratio of one). b) IL-8 secretion was measured 48 h after transfection (n=6).
Discussion
In the present study, using freshly obtained human nasal epithelial cells, it has been demonstrated that the elevated IL‐8 secretion produced by CF cells in the presence of P. aeruginosa is a direct consequence of increased bacterial binding. No increase in basal NF‐κB activity could be detected in primary CF nasal epithelial cells or in CF mice. NF‐κB activity and IL-8 secretion varied greatly between CF cell lines and neither was consistently increased, nor did they correlate with each other. Overexpression of ΔF508 and G551D mutant CFTR increased p50 activity in Cos‐7 cells when compared to overexpression of wild-type CFTR; however, this increase was not accompanied by an increase in IL‐8 secretion. Each of these studies has its limitations, but taken together the present findings lend support to the view that inflammation in CF relates predominantly to the bacterial burden in the airways of these patients.
There are substantial clinical data supporting the role of micro-organisms as the key initiator of CF inflammation 22, 23. Many pathogens are implicated in bronchial infection in CF including Haemophilius influenzae, Staphylococcus aureus, P. aeruginosa and respiratory syncytial virus (RSV). Each of the bacteria, and in particular P. aeruginosa, bind to asialo-GM1 receptors via pili, inducing subsequent IL‐8 production; no IL‐8 upregulation is observed using unpiliated P. aeruginosa. Thus, these findings are in keeping with the authors' suggestion that the characteristic CF inflammation is linked to the increase in P. aeruginosa adherence. The increased numbers of asialo-GM1 receptors on CF cells may be a direct result of a CF mutation-related sialylation defect 24. Thus, a reduction of P. aeruginosa adherence to nasal epithelial cells has been shown following transfection of wild-type CFTR 17.
Whilst P. aeruginosa adherence clearly triggers CF inflammation, other clinical studies suggest that inflammation may precede bacterial colonization. This is based on the detection in bronchoalveolar lavage of inflammatory cells and IL-8 in the absence of demonstrable micro-organisms 7, 8. An alternative explanation for these findings is that the inflammatory response has been successful in eradicating the organism which initially induced it 25. Several laboratory studies have also suggested a role for mutant CFTR directly inducing inflammation 10, 26. In keeping with the clinical studies showing normal IL‐8 levels after resolution of infection, no evidence for elevated IL‐8 in unstimulated primary CF nasal epithelial cells could be found. Of nasal swabs from 12 of the CF subjects taken to assess bacterial colonization, one was positive for S. aureus, whilst the remaining 11 showed no evidence of bacterial growth. Furthermore, no increase in NF-κB activation in CF, either in human nasal epithelium or in tissues obtained from ΔF508 CF mice could be detected. Clearly, one limitation of the whole lung EMSA analysis is the dilution effect of nonepithelial cells which are unlikely to express CFTR. However, the similarity of findings in the human epithelial cells support these data. Consistent with data published by DiMango et al. 10 increased NF‐κB activity and IL‐8 secretion in ΣCFTE29o- cells was also detected when compared to three wild-type cell lines. However, measurements in two other CF cell lines (CFSMEo-, CFNPE9o-) showed that an increase of NF-κB and IL‐8 was not a consistent characteristic of such immortalized CF cell lines. Indeed, all combinations of elevated or reduced NF‐κB activation and changes in IL‐8 could be demonstrated. Whilst the ΔF508 homozygous line (ΣCFTE29o-) produced higher levels of IL‐8 than the other CF cell lines, a recent paper has demonstrated a similar variability, including that between two ΔF508 homozygous cell lines 27.
Baeuerle et al. 28 have suggested that endoplasmic reticulum (ER) retention of proteins such as mutant CFTR may lead to activation of NF-κB and hence expression of pro-inflammatory cytokines. Cooling of ΔF508 CF cells has been shown to promote trafficking of ΔF508 CFTR to the apical membrane. A recent report has suggested that such a reduction in temperature also reduces NF‐κB activity and IL‐8 secretion 10. However, cooling is likely to affect a number of cellular processes, and the present study followed this up by promoting CFTR trafficking using trimethylamine N‐oxide (TMAO), which increases ΔF508 trafficking to the apical membrane in ΣCFTE29o- cells. TMAO could restore cyclic adenosine monophosphate (cAMP)-dependent chloride efflux, but did not alter NF‐κB activity and IL‐8 secretion in these cells (data not shown), consistent with the present data, showing lack of effect of different CFTR mutations. Clearly, however, these results may be interpreted as TMAO inducing a low level of trafficking sufficient to restore chloride function, but leaving the vast majority of mutant CFTR mistrafficked.
In an attempt to further determine the effect of ER overload, the present study transiently overexpressed wild-type, ΔF508 or G551D CFTR protein in Cos‐7 cells. These were chosen for their relatively high transfection efficiency and also because these cells have the capability to respond to an inflammatory stimulus, such as TNF‐α, with increased IL‐8 secretion (data not shown). A significant increase in a p50 containing protein/DNA complex (complex II) was detected after ΔF508 or G551D overexpression when compared to overexpression of the wild-type protein. However, the increased p50 activity was not accompanied by an increase in IL‐8 secretion. At present it is uncertain to what extent p50 homo- or heterodimers contribute to IL‐8 regulation. Zhu et al. 29 have demonstrated that p65/65 homodimers are responsible for the regulation of IL‐8 secretion in human nasal epithelial cells. However, other studies indicate that IL‐8 secretion is regulated through p50/p65 heterodimers 30 and it is likely that regulation varies between cell types.
The present findings are in keeping with the view that increased bacterial adherence contributes significantly to the inflammatory response in cystic fibrosis airways. Host inflammatory responses in cystic fibrosis may be considered in two phases during disease progression. The first phase is characterized by increased bacterial adherence and a preserved capacity for elimination of pathogens, associated with enhanced inflammatory markers as witnesses of recent infection. However, the effects of a chronic inflammatory response, and the continued presence of bacteria (related to increased adherence, excess mucus and probably many other factors), lead to the second phase where the inflammatory process is no longer sufficiently effective to produce bacterial clearance. The present data cannot rule out a direct contribution of the mutant protein on inflammation, but it is suggested that this may be a less significant factor. If these views are correct, novel treatments aimed at reducing bacterial adherence would be expected to play an important role early in life in cystic fibrosis patients. Reducing inflammation at this time may prove harmful, by reducing the appropriate host response to an increased burden. The impact of such treatments later in life will clearly depend on the balance between “useful” and “inappropriate” inflammation. It is likely that only trials of anti-inflammatory treatment can distinguish these possibilities.
Acknowledgments
The authors thank the volunteers who provided the nasal samples, and the staff of the Royal Brompton Hospital Cystic Fibrosis Clinic for their help. In addition, the authors thank T. Pitt for providing the Pseudomonas aeruginosa bacterial strain, D. Gruenert for the cell lines, L. Huang for the DC-Cholesterol/dioleylphosphatidylethanolamine (DOPE) and J. Rommens for the cystic fibrosis transmembrane conductance regulator (CFTR) plasmids.
- Received April 3, 2000.
- Accepted July 31, 2000.
- © ERS Journals Ltd
References









